
Строение и функции белков
Урок изучения нового материала в 10-м классе. Данный материал учащиеся уже изучали в 9 классе, поэтому некоторые понятия им уже известны. Соответственно с ребятами ведется диалог о строении и функциях белков. С помощью учителя учащиеся узнают о классификации ферментов.
Для того, чтобы активизировать деятельность учащихся на уроке, приводятся интересные факты о белках, которые помогают ребятам и нацеливают их на дальнейшее усвоение нового материала. Так же для этих целей предлагается провести лабораторную работу. На данном уроке основная масса изучаемого материала записывается в виде таблиц, схемы, которые учитель строит в ходе урока вместе и учениками. Качество изучаемого материала проверяется в виде фронтального опроса. Урок рассчитан как на детей-аудиалов, так и визуалов.
Цель урока: дать представление о строении и функции белков.
Задачи: продолжить расширение и углубление знаний важнейших органических
веществах клетки на основе изучения строения и функции белков, сформировать
знания функциях белков и их важнейшей роли в органическом мире, продолжить
формирование умения выявлять связи между строением и функциями веществ.
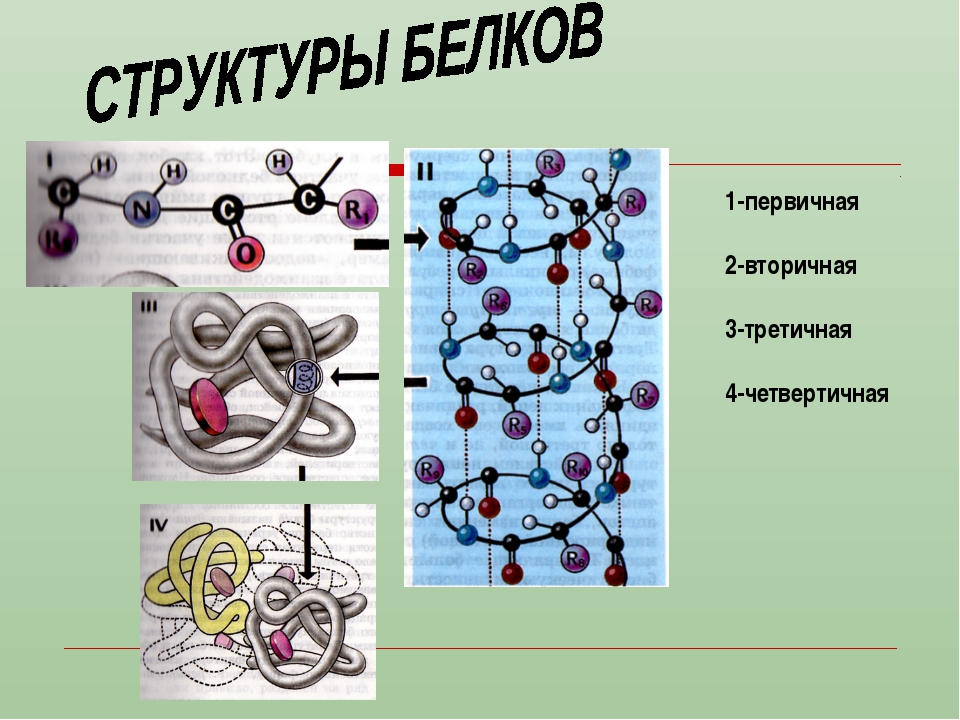
Основные понятия: белки, протеины, протеиды, пептид, пептидная связь, простые и сложные белки, первичная, вторичная, третичная и четвертичная структуры белков денатурация.
Средства обучения: таблицы по общей биологии, иллюстрирующие строение молекул белков; лабораторное оборудование для проведения лабораторной работы “Расщепление пероксида водорода с помощью ферментов, содержащихся в плетках листа элодеи”.
Ход урока
I. Изучение нового материала.
1. Рассказ учителя (или фрагмент лекции) об особенностях строения молекул белков как биополимеров, состоящих из большого количества разных аминокислот, между которыми происходит полимеризация на основе пептидной связи. Зарисовка и запись на доске и в тетрадях учащихся.
2. Самостоятельное изучение учащимися текста учебника (С.42) о классификации белков.
3. Беседа об уровнях организации белковой молекулы и химической основы
каждого из четырех уровней (структур) этой молекулы, о денатурации как утрате
белковой молекулы своей природной структуры.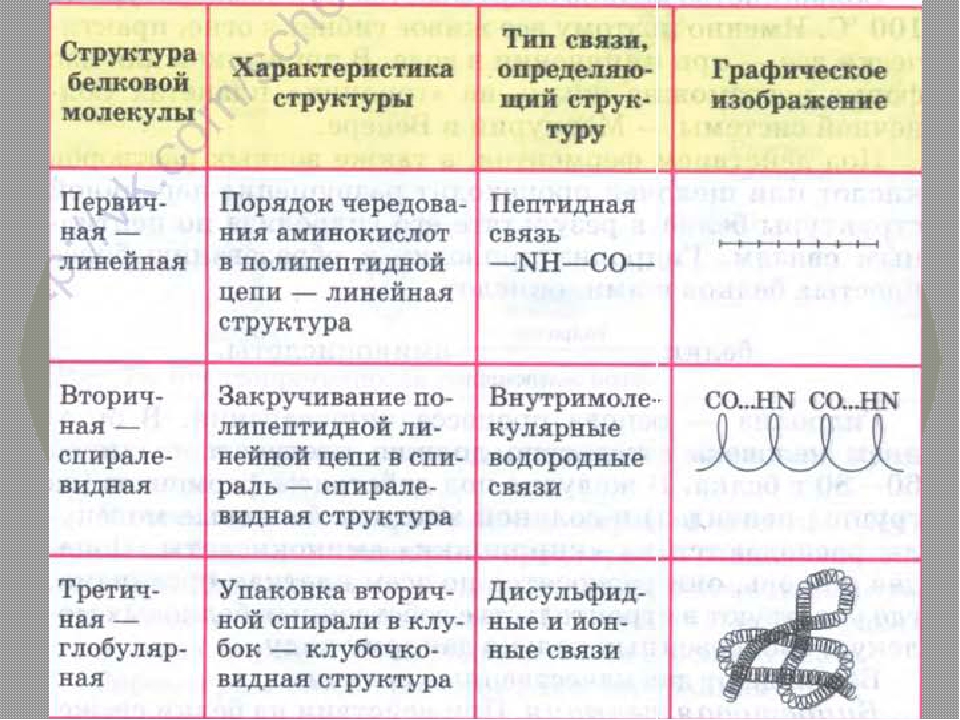
Структура белковой молекулы.
| Структура белка | Характеристика | Тип связи | |
| Первичная | Линейная структура – последовательность аминокислот в полипептидной цепи, которая определяет все другие структуры молекулы, а также свойства и функции белка. | Пептидная. | |
| Вторичная | Закручивание полипептидной цепи в
спираль или складывание в “гармошку”. |
Водородные связи. | |
| Третичная | Глобулярный белок: упаковка вторичной
структуры в глобулу; фибриллярный белок: несколько вторичных структур, уложенных параллельными слоями, или скручивание нескольких вторичных структур наподобие каната в суперспираль. |
Ионные, водородные, дисульфидные, гидрофобные. | |
| Четвертичная | Встречается редко. Комплекс из нескольких третичных структур органической природы и неорганическое вещество, например, гемоглобин. | Ионные, водородные, гидрофобные. |
4. Рассказ учителя о многообразии функций белков с краткой записью в тетрадях
сущности функций: структурной, ферментативной, транспортной, защитной,
регуляторной, энергетической, сигнальной.
Рассказ учителя о многообразии функций белков с краткой записью в тетрадях
сущности функций: структурной, ферментативной, транспортной, защитной,
регуляторной, энергетической, сигнальной.
5. Лабораторная работа “Расщепление пероксида водорода с помощью ферментов, содержащихся в клетках листа элодеи”.
Ход работы:
а. Приготовьте микропрепарат листа элодеи и рассмотрите его под микроскопом.
б. Капните на микропрепарат немного пероксида водорода и еще раз рассмотрите, в
каком состоянии находятся клетки листа элодеи.
в. Объясните, с чем связано выделение пузырьков из теток листа, что это за газ,
на какие вещества может расщепиться пероксид водорода, какие ферменты участвуют
в этом процессе?
г. Капните каплю пероксида на предметное стекло и, рассмотрев его под
микроскопом, опишите наблюдаемую, картину. Сравните состояние пероксида водорода
в листе элодеи и на стекле, сделайте выводы.
По завершении лабораторной работы следует провести беседу о биохимических реакциях, протекающих при участии белковых катализаторов-ферментов как основе жизнедеятельности клеток и организмов.
Химические свойства белков обусловлены их различным аминокислотным составом. Существуют белки хорошо растворимые в воде и совершенно нерастворимые, химически активные и устойчивые к действию различных агентов, способные укорачиваться и растягиваться и т. д.
Под влиянием различных факторов – высокой температуры, действия химических
веществ, облучения, механического воздействия – может произойти разрушение
структур белковой молекулы. Нарушение природной структуры белка называется
денатурацией. Если воздействие перечисленных факторов было недолгим и несильным,
то белок может вернуть свою природную структуру – обратимая денатурация (ренатурация),
если же воздействие было долгим или сильным, то происходит нарушение не только
третичной и вторичной структур, но и первичной – необратимая денатурация (рис.
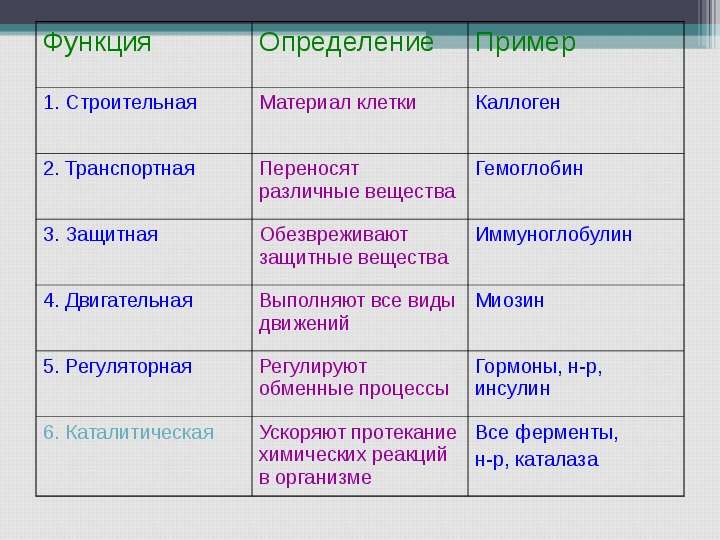
Функции белков.
| Функция | Характеристика |
| 1. Строительная (структурная). | Входят в состав клеточных мембран и органоидов клетки (липопротеиды и гликопротеиды), участвуют в образовании стенок кровеносных сосудов, хрящей, сухожилий (коллаген) и волос (кератин). |
| 2. Двигательная | Обеспечивается сократительными белками (актин и миозин), которые обуславливают движение ресничек и жгутиков, сокращение мышц, перемещение хромосом при делении клетки, движение органов растений. |
| 3. Транспортная. | Связывают и переносят с током крови
многие химические соединения, например, гемоглобин и миоглобин
транспортируют кислород, белки сыворотки крови переносят гормоны, липиды
и жирные кислоты, различные биологически активные вещества. |
| 4. Защитная. | Выработка антител (иммуноглобулинов) в ответ на проникновение в нее чужеродных веществ (антигенов), которые обеспечивают иммунологическую защиту; участие в процессах свертывания крови (фибриноген и протромбин). |
| 5, Сигнальная (рецепторная). | |
| 6. Регуляторная. | Белки-гормоны оказывают влияние на
обмен веществ, т. е. обеспечивают гомеостаз, регулируют рост,
размножение, развитие и другие жизненно важные процессы. Например,
инсулин регулирует уровень глюкозы в крови, тироксин – физическое и
психическое развитие и т.д. е. обеспечивают гомеостаз, регулируют рост,
размножение, развитие и другие жизненно важные процессы. Например,
инсулин регулирует уровень глюкозы в крови, тироксин – физическое и
психическое развитие и т.д. |
| 7. Каталитическая (ферментативная). | Белки-ферменты ускоряют биохимические процессы в клетке. |
| К. Запасающая | Резервные белки животных: альбумин (яйца) запасает воду, ферритин – железо в клетках печени, селезенки; миоглобин – кислород в мышечных волокнах, казеин (молоко) и белки семян – источник питания для зародыша. |
| 9. Пищевая (основной источник аминокислот). | Белки пищи – основной источник
аминокислот (особенно незаменимых) для животных и человека; казеин
(белок молока) – основной источник аминокислот для детенышей
млекопитающих. |
| 10. Энергетическая. |
Ферменты (энзимы) – это специфические белки, которые присутствуют во всех живых организмах и играют роль биологических катализаторов.
Химические реакции в живой клетке протекают при умеренной температуре,
нормальном давлении и нейтральной среде. В таких условиях реакции синтеза или
распада веществ протекали бы очень медленно, если бы не подвергались воздействию
ферментов. Ферменты ускоряют реакцию без изменения ее общего результата за счет
снижения энергии активации. Это означает, что в их присутствии требуется
значительно меньше энергии для придания реакционной способности молекулам,
которые вступают в реакцию. Ферменты отличаются от химических катализаторов
высокой степенью специфичности, т. е. фермент катализирует только одну реакцию
или действует только на один тип связи. Скорость ферментативных реакций зависит
от многих факторов – природы и концентрации фермента и субстрата, температуры,
давления, кислотности среды, наличия ингибиторов и т.д.
Ферменты отличаются от химических катализаторов
высокой степенью специфичности, т. е. фермент катализирует только одну реакцию
или действует только на один тип связи. Скорость ферментативных реакций зависит
от многих факторов – природы и концентрации фермента и субстрата, температуры,
давления, кислотности среды, наличия ингибиторов и т.д.
Классификация ферментов.
| Группа | Катализируемые реакции, примеры |
| Оксидоредуктазы. | Окислительно-восстановительные
реакции: перенос атомов водорода (Н) и кислорода (О) или электронов от
одного вещества к другому, при этом окисляется первый и
восстанавливается второй. Участвуют во всех процессах биологического
окисления, например, вдыхании: АН + В А
ВН (окисленный)
или А + О АО
(восстановленный). |
| Трансферазы. | Перенос группы атомов (метильной,
ацильной, фосфатной или аминогруппы) от одного вещества к другому.
Например, перенос остатков фосфорной кислоты от АТФ на глюкозу или
фруктозу под действием фототрансфераз: АТФ + глюкоза глюкозо-6-фосфат + АДФ. |
| Гидролазы. | Реакции расщепления сложных
органических соединений на более простые путем присоединения молекул
воды в месте разрыва химической связи (гидролиз). Например, амилаза (гидролизирует
крахмал), липаза (расщепляет жиры), трипсин (расщепляет белки) и др.: АВ + Н20 АОН + ВН. |
| Лиазы | Негидролитическое присоединение к
субстрату или отщепление от него группы атомов. При этом могут
разрываться связи С-С, C-N, С-О, C-S. Например, декарбоксилаза отщепляет
карбоксильную группу: При этом могут
разрываться связи С-С, C-N, С-О, C-S. Например, декарбоксилаза отщепляет
карбоксильную группу: |
| Изомеразы | Внутримолекулярные перестройки,
превращение одного изомера в другой (изомеризация): глюкозо-6-фосфат глюкозо-1-фосфат. |
| Лигазы (синтетазы) | Реакции соединения двух молекул с
образованием новых связей С–О, С–S, С–N, С–С, с использованием энергии
АТФ. Например, фермент валин-тРНК-синтетаза, под действием которого
образуется комплекс валин– тРНК: АТФ + валин + тРНК АДФ + Н3Р04 + валин-тРНК. |
Механизм действия фермента представлен на рис. 4. В молекуле каждого фермента
имеется активный центр – это один или более участков, в которых происходит
катализ за счет тесного контакта между молекулами фермента и специфического
вещества (субстрата). Активным центром выступает или функциональная группа
(например, ОН-группа), или отдельная аминокислота. Активный центр может
формироваться связанными с ферментом ионами металлов, витаминами и другими
соединениями небелковой природы – коферментами или кофакторами. Форма и
химическое строение активного центра таковы, что с ним могут связываться только
определенные субстраты в силу их идеального соответствия (комплементарности)
друг другу.
Активным центром выступает или функциональная группа
(например, ОН-группа), или отдельная аминокислота. Активный центр может
формироваться связанными с ферментом ионами металлов, витаминами и другими
соединениями небелковой природы – коферментами или кофакторами. Форма и
химическое строение активного центра таковы, что с ним могут связываться только
определенные субстраты в силу их идеального соответствия (комплементарности)
друг другу.
Молекула фермента изменяет глобулярную форму молекулы субстрата. Молекула субстрата, присоединяясь к ферменту, тоже в определенных пределах изменяет свою конфигурацию для увеличения реакционности функциональных групп центра.
На заключительном этапе химической реакции фермент-субстратный комплекс распадается с образованием конечных продуктов и свободного фермента. Освободившийся при этом активный центр может принимать новые молекулы субстрата.
II. Обобщающая беседа об основополагающей роли белков как самых
необходимых химических соединений для жизней деятельности всего живого на Земле.
III. Закрепление знаний в процессе беседы с помощью следующих вопросов:
- Какие органические вещества клетки можно назвать самыми важными?
- Каким образом создается бесконечное разнообразие белков?
- Что собой представляют мономеры биополимера белка?
- Как формируется пептидная связь?
- Что собой представляет первичная структура белка?
- Каким образом происходит переход первичной структуры молекул белка во вторичную, а затем– в третичную и четвертичную?
- Какие функции могут выполнять белковые молекулы?
- Чем обусловлено многообразие функций белковых молекул?
- Приведите примеры белков, выполняющих самые разные функции. При ответе можно использовать следующую схему:
Биологические функции белков.
Это интересно.
Многие молекулы очень велики и по длине, и по молекулярной массе. Так,
молекулярная масса инсулина – 5700, белка-фермента рибонуклеазы – 127 ООО,
яичного альбумина – 36 ООО, гемоглобина – 65 ООО. В состав различных белков
входят самые разные аминокислоты. Набор всех двадцати видов аминокислот
содержит: казеин молока, миозин мышц и альбумин яйца. В белке-ферменте
рибонуклеазе – 19, в инсулине – 18 аминокислот. Коллективу ученых под
руководством академика Ю.А. Овчинникова удалось расшифровать сложную структуру
белка родопсина, ответственного за процесс зрительного восприятия.
Так,
молекулярная масса инсулина – 5700, белка-фермента рибонуклеазы – 127 ООО,
яичного альбумина – 36 ООО, гемоглобина – 65 ООО. В состав различных белков
входят самые разные аминокислоты. Набор всех двадцати видов аминокислот
содержит: казеин молока, миозин мышц и альбумин яйца. В белке-ферменте
рибонуклеазе – 19, в инсулине – 18 аминокислот. Коллективу ученых под
руководством академика Ю.А. Овчинникова удалось расшифровать сложную структуру
белка родопсина, ответственного за процесс зрительного восприятия.
Кровь осьминогов, моллюсков и пауков имеет голубой цвет, потому что переносчиком кислорода у них служит не красный гемоглобин, содержащий атомы железа, а гемоцианин с атомами меди.
Почти половина необходимых нам белков, углеводов, 70–80% витаминов, значительное количество минеральных солей, аминокислот и других питательных элементов содержится в хлебе.
Американские ученые выделили из растения (семейство Пентадипландовых),
произрастающего в Западной Африке, белок, который слаще сахара в 2 тыс. раз.
Этот шестой известный науке сладкий белок, названный бразеином, содержится в
плодах, которые с большой охотой поедают местные обезьяны. Биохимики
расшифровали строение молекул сладкого белка, в каждой из них содержится 54
аминокислотных остатка.
раз.
Этот шестой известный науке сладкий белок, названный бразеином, содержится в
плодах, которые с большой охотой поедают местные обезьяны. Биохимики
расшифровали строение молекул сладкого белка, в каждой из них содержится 54
аминокислотных остатка.
IV. Домашнее задание: Изучить § 11, ответить на вопросы на с. 46. Приготовить сообщения или рефераты на темы: “Белки – биополимеры жизни”, “Функции белков – основа жизнедеятельности каждого организма на Земле”, “Денатурация и ренатурация, ее практическое значение”, “Многообразие ферментов, их роль в жизнедеятельности клеток и организмов” и др.
Используемые ресурсы:
- Каменский А.А.Общая биология 10–11: учеб.для общеобразоват. учреждений.– М.:Дрофа, 2006.
- Козлова Т.А. Тематическое и поурочное планирование по биологии к
учебнику А.А.Каменского и др. “Общая биология 10–11”. – М.: Издательство
“Экзамен”, 2006.

- Биология. Общая биология. 10–11 классы: рабочая тетрадь к учебнику Каменского А.А. и др. “Общая биология 10–11”– М.: Дрофа, 2011.
- Кириленко А.А. Молекулярная биология. Сборник заданий для подготовки к ЕГЭ: уровни А,В,С: учебно-методическое пособие. – Ростов н/Д: Легион, 2011.
Обзор архитектуры AlphaFold 2 / Хабр
В данном обзоре мы подробно рассмотрим нейронную сеть AlphaFold 2 от компании DeepMind, с помощью которой недавно был совершен прорыв в одной из важных задач биологии и медицины: определении трехмерной структуры белка по его аминокислотной последовательности.
В первых трех разделах обзора описывается задача, формат входных данных и общая архитектура AlphaFold 2. Далее, начиная с раздела «Input feature embeddings», описываются детали архитектуры. В разделе «Резюме» кратко суммируется основная информация из обзора.
В научной статье, опубликованной в Nature, и дополнительных материалах к ней, авторы используют название «AlphaFold» без цифры 2, и мы также будем его придерживаться.
Белки и их структуры
Белки – это органические молекулы, структура которых показана на рис. 1. Символом R обозначены аминокислотные остатки, которые могут быть 20 разных типов. Таким образом, белок можно закодировать строкой, записанной алфавитом из 20 символов.
Рис. 1. Структура белка.Белки сворачиваются в структуру за счет различных взаимодействий между атомами (водородные связи, ковалентные связи и др.). Структура белка определяется его аминокислотной последовательностью и, в свою очередь, определяет свойства этого белка в живом организме.
Задача, которую решает AlphaFold, заключается в предсказании структуры белка по его аминокислотной последовательности. Экспериментальное определение структуры белков является очень трудоемким. Решение этой задачи с помощью физического моделирования требует огромных вычислительных ресурсов. Проблема усугубляется тем, что в процессе сворачивания белка часто участвуют другие белки.
Каждые 2 года организуется соревнование CASP (Critical Assessment of protein Structure Prediction), в котором научные группы соревнуются в точности предсказания структур белков. Для оценки точности каждый раз используется новый набор белков, структуры которых уже были получены экспериментально, но еще не были опубликованы.
Для оценки точности каждый раз используется новый набор белков, структуры которых уже были получены экспериментально, но еще не были опубликованы.
В 2020 году DeepMind с их нейронной сетью AlphaFold 2 выиграла соревнование CASP14, достигнув беспрецедентного уровня точности (рис. 2). DeepMind также выложили на YouTube видео об этом историческом успехе, которое рекомендую посмотреть. Об архитектуре AlphaFold версии 2 и пойдет речь в этом обзоре.
Рис. 2. Максимальная точность по метрике GDT, достигнутая в ходе соревнований CASP в разные годы (1994-2020).Предсказание структуры на основе эволюционного сходства
Аминокислотная последовательность белков меняется в процессе эволюции. Лишь часть аминокислот в цепочке влияет на структуру белка: мутации в этих местах скорее всего приведут к неправильному сворачиванию белка и утрате им своих полезных свойств. В результате организм с большой вероятностью отсеивается естественным отбором. Поэтому в ходе эволюции мутации в основном накапливаются в тех местах белка, которые не оказывают влияния на его структуру.
Если мы возьмем белок, выполняющий одну и ту же функцию в разных живых организмах, то увидим различия, накопившиеся в ходе эволюции от общего предка. Такое сопоставление называется множественным выравниванием последовательностей (multiple sequence alignment, MSA). Пример показан на рис. 3. Каждая строка – вид организма, столбец – код аминокислоты. Иногда в ходе эволюции аминокислоты могут удаляться или добавляться в белок. Чтобы было возможно выравнивание, удаленные аминокислоты в MSA обозначаются дефисом.
Рис. 3. Пример таблицы MSA.Например, мы видим, что аминокислота на позиции 22 (лизин, символ «K») одинакова у всех организмов в таблице, тогда как аминокислота на позиции 23 сильно варьируется. Это говорит о том, что аминокислота К в данной позиции важна для сохранения структуры белка.
Мутации в местах, определяющих структуру белка, не только редки, но обычно происходят «парами»: то есть сразу два аминокислотных остатка, которые находятся в контакте друг с другом, мутируют так, что контакт сохраняется, а значит сохраняется структура белка и его свойства (рис. 4). В результате организм получается жизнеспособным и не отсеивается естественным отбором.
4). В результате организм получается жизнеспособным и не отсеивается естественным отбором.
Таблицу MSA можно составить для любого белка, найдя в базе данных наиболее схожие и эволюционно близкие к нему белки. Коррелирующие элементы последовательности вероятно контактируют, часто меняющиеся – не влияют на структуру. Поэтому таблицу MSA можно использовать при предсказании трехмерной структуры белка. Более того, для многих белков уже известны трехмерные структуры. Если в MSA найдутся белки с известными структурами, то по их структуре можно попытаться восстановить структуру исследуемого белка.
Предварительная обработка данных
Формат входных данных
Белок, для которого требуется определить структуру, будем называть целевым белком. Его аминокислотная последовательность представлена в виде строки из символов. Для целевого белка выполняется поиск в базе данных и составляется таблица MSA. Также происходит поиск шаблонов (templates): нескольких наиболее похожих белков с известной структурой. Каждый шаблон представлен в виде координат атомов в пространстве. Как вариант, может не быть ни одного шаблона. Таким образом, входными данными являются:
Каждый шаблон представлен в виде координат атомов в пространстве. Как вариант, может не быть ни одного шаблона. Таким образом, входными данными являются:
Целевой белок
Таблица MSA
Шаблоны (опционально)
Целевой белок
Целевой белок состоит из последовательности аминокислотных остатков (residues), для которых нужно определить пространственные положения.
Для целевого белка выполняется one-hot кодирование (20 аминокислот + «неизвестно»). Таким образом, аминокислотная последовательность длиной превращается в массив из нулей и единиц размером .
Обрезка целевого белка и таблицы MSA
По оси, соответствующей номеру аминокислоты, целевой белок и таблица MSA обрезаются до фиксированной длины (выбирается случайный участок таблицы). На разных этапах обучения AlphaFold размер вырезаемого участка равен 256 и 384.
Создается массив , который хранит для целевого белка позиции аминокислот до обрезки.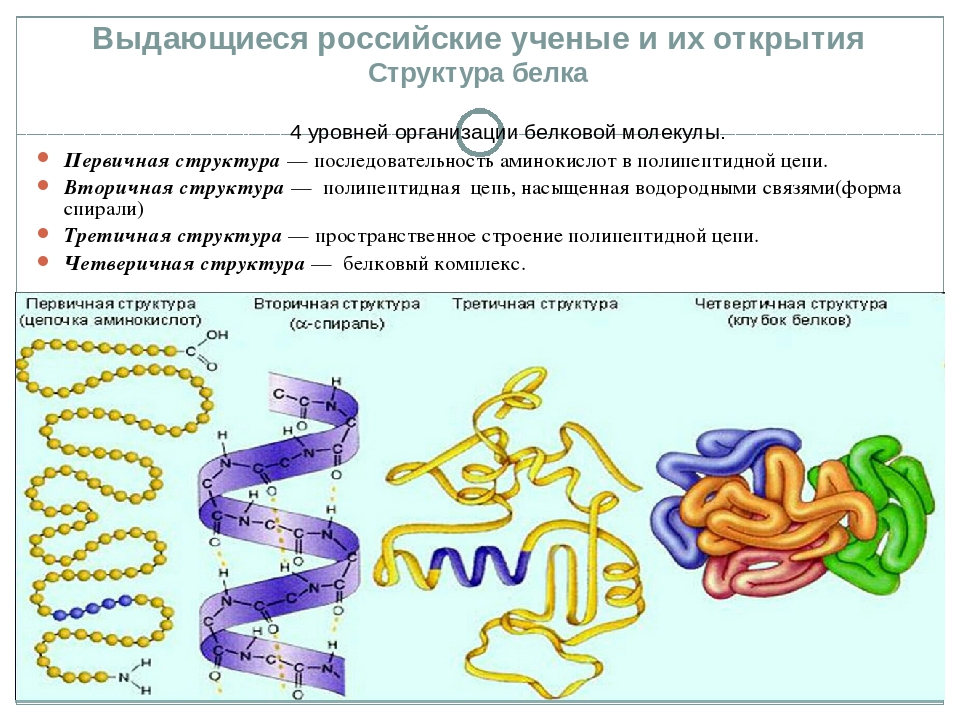 Например, если мы обрезали белок с 20 по 40 позицию, то массив будет состоять из чисел [20, 21, …, 39].
Например, если мы обрезали белок с 20 по 40 позицию, то массив будет состоять из чисел [20, 21, …, 39].
Подробнее см. Supplementary Material, раздел 1.2.8
Примечание. Здесь остается вопрос: насколько точно можно предсказать структуру белка, если обрезать его часть? В работе я не нашел освещения этого вопроса, но точность AlphaFold говорит сама за себя. Мне удалось найти таблицу распределения белков по длинам аминокислотных последовательностей (рис. 5).
Рис. 5. Распределение белков по длинам аминокислотных последовательностей в базе данных PDB30.Кластеризация и маскирование таблицы MSA
Сложность вычислений и объем требуемой памяти в AlphaFold квадратично зависит от количества последовательностей в MSA, и лишь линейно от длины последовательности, поэтому количество последовательностей в MSA желательно уменьшить. Можно было бы выбирать случайную подвыборку последовательностей, но авторы предлагают другой подход: последовательности объединяются в кластеры.
В качестве метрики расстояния выбирается расстояние Хэмминга между последовательностями: число позиций, в которых аминокислоты различаются. Случайная подвыборка последовательностей выбирается в качестве центров кластеров, для всех остальных последовательностей ищется ближайший кластер.
Также 15% аминокислот в MSA «маскируется», то есть заменяется на случайную аминокислоту или на символ [MASK], для которого также выделяется бит в one-hot кодировании. Одна из задач сети AlphaFold 2 в ходе обучения состоит в том, чтобы предсказать замененные аминокислоты. Это работает аналогично задаче «masked language model» в языковой модели BERT.
Подробнее см. Supplementary Material, раздел 1.2.7
Если количество кластеров равно , а длина последовательности равна , то данные собираются в массив размером (, , 49). Для каждого , вектор размерностью 49 является конкатенацией one-hot кодирования -й аминокислоты центра -го кластера, распределения по аминокислотам для всего -го кластера и некоторых дополнительных данных.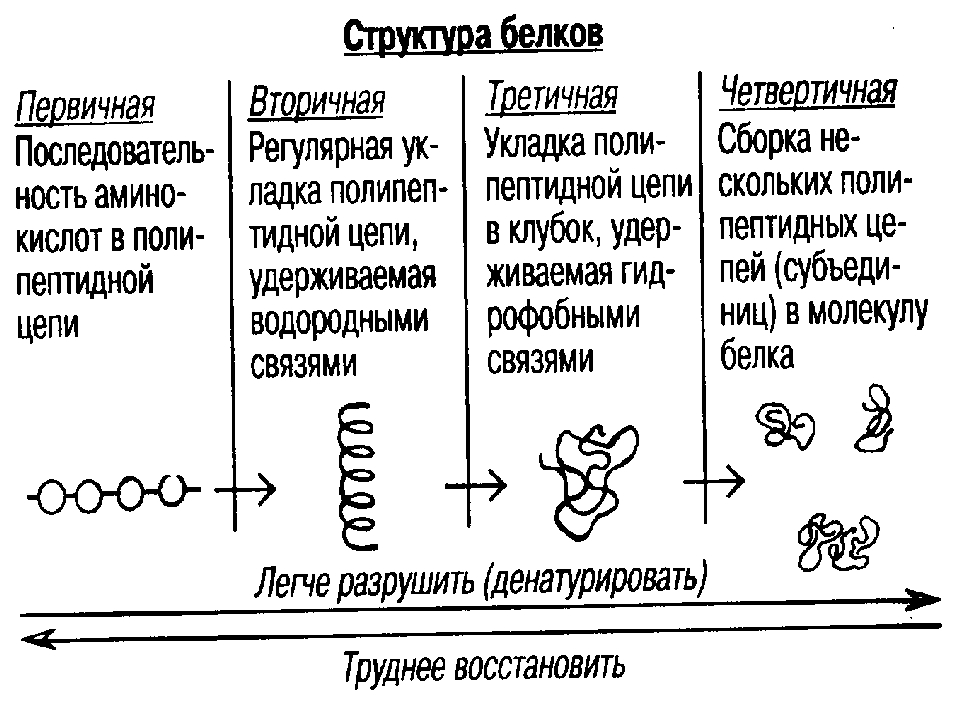
Таким образом, массив содержит информацию о таблице MSA, после кластеризации, маскирования и one-hot кодирования.
На этом, однако, сложности не заканчиваются. Создается еще один дополнительный массив , состоящий из дополнительного набора последовательностей из MSA, не включенных в кластеры. Аминокислоты также кодируются one-hot кодированием, добавляются некоторые дополнительные данные. В результате массив имеет размер (, , 25).
Подробнее см. Supplementary Material, раздел 1.2.9
Данные о шаблонах
Исходными данными служат координаты атомов -углерода (или -углерода для аминокислоты глицин) (рис. 6).
Рис. 6. Атомы alpha- и beta-углерода в молекуле белка.Создается массив попарных расстояний (авторы используют термин «дистограмма», англ. distogram) между атомами -углерода. Этот массив инвариантен к сдвигу, повороту и отражению системы координат. Далее выполняется дискретизация каждого расстояния между 3. 25 Å и 50.75 Å с 39 возможными значениями (последнее значение – «50.75 Å или больше»). После дискретизации выполняется one-hot кодирование.
25 Å и 50.75 Å с 39 возможными значениями (последнее значение – «50.75 Å или больше»). После дискретизации выполняется one-hot кодирование.
Массив состоит из полученной дистограммы и некоторых дополнительных данных. Этот массив имеет размер (, , , 88), где – количество шаблонов.
Массив содержит информацию об аминокислотах, из которых состоят шаблоны, и об углах, под которыми соединены атомы в цепочке.
Подробнее см. Supplementary Material, раздел 1.2.9
Результаты обработки
В результате предварительной обработки данных мы получили 6 массивов. Их размеры и описания суммаризированы ниже.
– данные об аминокислотах целевого белка
– данные о том, какую часть целевого белка мы обрезали
– данные о кластеризованной таблице MSA
– данные о дополнительных последовательностях в MSA
– данные о шаблонах, в т. ч. попарные расстояния между атомами в каждом шаблоне
– данные о шаблонах: аминокислоты и углы между атомами в каждом шаблоне
Архитектура AlphaFold
В этом разделе мы рассмотрим основные вычислительные блоки, из которых состоит архитектура AlphaFold версии 2. Рассмотрим назначение каждого блока и формат передаваемых между ними данных, но пока не будем детально рассматривать внутреннее устройство блоков.
Рассмотрим назначение каждого блока и формат передаваемых между ними данных, но пока не будем детально рассматривать внутреннее устройство блоков.
Модель AlphaFold способна выдавать ответ (трехмерную структуру белка) за один end-to-end запуск. Но авторы обнаружили, что можно улучшить качество предсказаний, запуская AlphaFold многократно (recycling iterations): каждую следующую итерацию модель использует выходные данные, полученные на предыдущей итерации, и обновляет предсказания. Это усложняет архитектуру, поэтому сначала рассмотрим более простой вариант без использования recycling iterations (Рис. 7).
Рис. 7. Упрощенный вариант AlphaFold без recycling iterations.Input feature embeddings
Первый блок AlphaFold принимает на вход 6 массивов, которые мы получили в результате предварительной обработки данных. Этот блок выполняет последовательность операций с обучаемыми весами, и его выходными данными являются три массива:
MSA representation, размером .
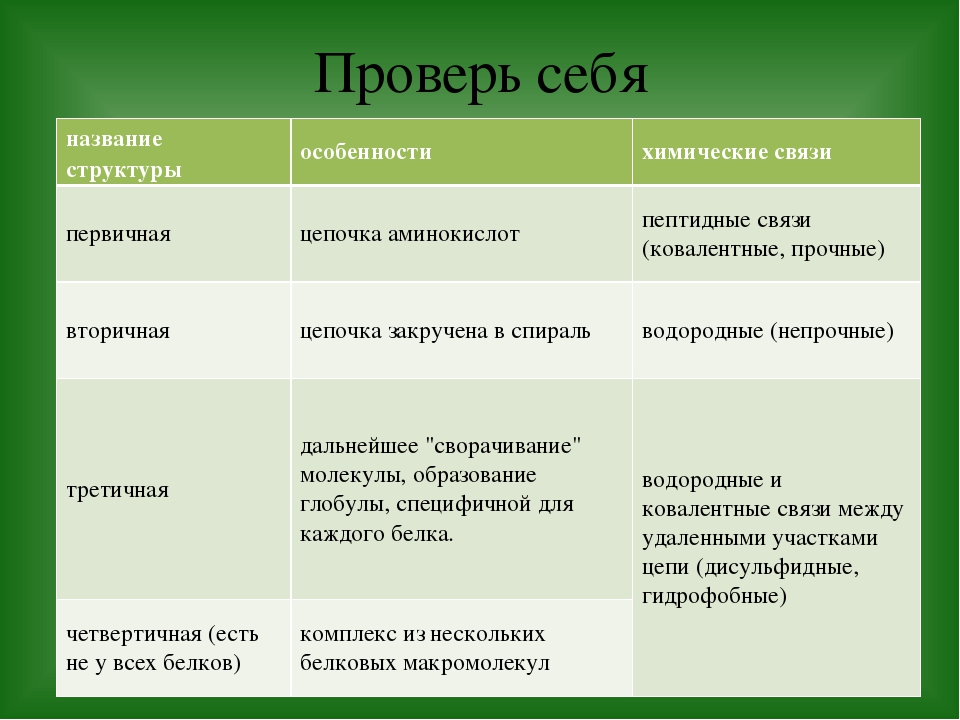 Вектора-эмбеддинги для каждой пары «позиция + номер кластера». Под «позицией» имеем в виду номер аминокислотного остатка.
Вектора-эмбеддинги для каждой пары «позиция + номер кластера». Под «позицией» имеем в виду номер аминокислотного остатка.Pair representation, размером . Вектора-эмбеддинги для каждой пары позиций.
Extra MSA representation, размером . Вектора-эмбеддинги для каждой пары «позиция + номер последовательности».
Это работает так же, как в задачах NLP (эмбеддинги слов). Входные данные, изначально представленные в понятном человеку формате, мы переводим в некий «внутренний формат», понятный только нейронной сети. Например, в массиве pair representation каждой паре позиций соответствует вектор размерностью 128.
Элемент массива pair representation является вектором-эмбеддингом пары позиций , или, иначе говоря, эмбеддингом ориентированного ребра, связывающего позиции и (рис. 8). При этом считается, что между каждой парой позиций есть два ориентированных ребра (полносвязный граф).
Рис. 8. Элемент pair representation как эмбеддинг направленного ребра.
Evoformer stack
Evoformer-блок – это вычислительный блок с обучаемыми весами, разработанный для архитектуры AlphaFold. Этот блок принимает на вход массивы pair representation и MSA representation и возвращает два массива таких же размеров. Внутри Evoformer-блока происходит обмен информацией между массивами и их обновление.
В первых 4 evoformer-блоках осуществляется обмен информацией между pair representation и extra MSA representation. В следующих 48 блоках осуществляется обмен информацией между pair representation и MSA representation.
Внутреннее устройство evoformer-блока мы разберем позднее. Пока достаточно сказать, что Evoformer-блок близок к трансформеру (Vaswani et al. 2017) и использует механизм self-attention.
Именно с помощью evoformer-блоков AlphaFold определяет трехмерную структуру белка. Полученный на выходе обновленный массив pair representation теперь содержит информацию о трехмерной структуре, и задача следующего блока – извлечь эту информацию и построить саму структуру в явном виде.
Примечание. Для построения трехмерной структуры (с точностью до отражения) достаточно иметь матрицу попарных расстояний между позициями, то есть массив размером (, ). Поскольку массив pair representation имеет в 128 раз больший размер, нейронной сети должно не составить труда закодировать в нем ту же информацию. Кроме того, в pair representation должна быть закодирована информация, позволяющая восстановить не только позиции атомов -углерода, но и позиции все остальных атомов углерода и азота в молекуле белка, а также оценить уверенность в предсказаниях.
Еще одним выходом Evoformer stack является массив single representation: для этого каждый вектор первой строки массива MSA representation, полученного на выходе, обрабатывается полносвязным слоем. Смысл массива single representation в том, что он кодирует информацию о каждом остатке в отдельности, а не о каждой паре остатков.
На схеме AlphaFold, которая была приведена выше, авторы для краткости не показали первые 4 evoformer-блока (extra evoformer stack).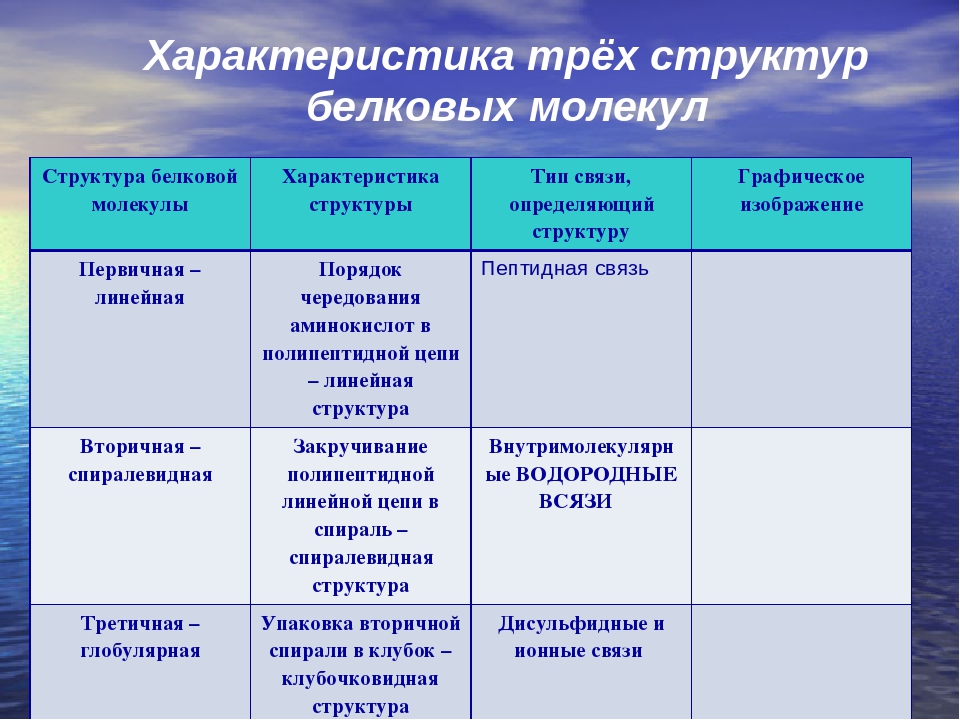 Более подробная схема показана рис. 9.
Более подробная схема показана рис. 9.
Structure module
После получения выходных данных evoformer stack работа с MSA-таблицей заканчивается. Блок structure module принимает на вход данные, полученные на выходе из evoformer stack:
single representation размером – вектора-эмбеддинги для каждой позиции
pair representation размером – вектора-эмбеддинги для каждой пары позиций
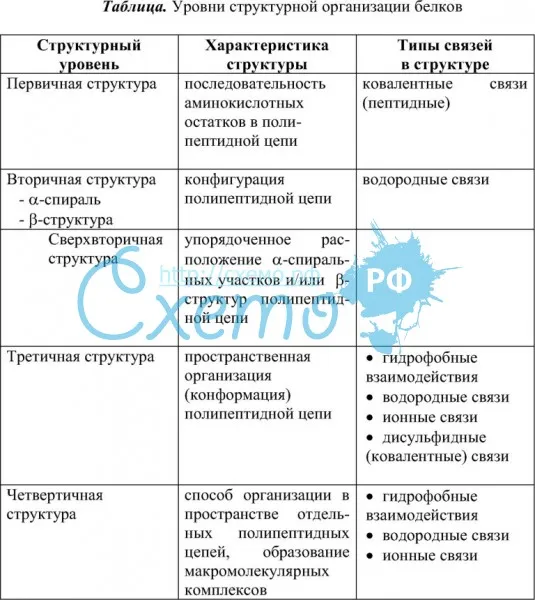
Предполагается, что в этих массивах уже закодирована информация о возможной структуре белка. Остается построить саму структуру. В задаче предсказания структуры белка выходные данные могут иметь разный формат, например:
Вариант 1. Координаты всех атомов белка. Систему координат можно выбрать произвольно, поэтому такое представление не единственно. Из-за этого не так просто подобрать подходящую функцию потерь.
Вариант 2. Массив попарных расстояний между атомами. Такое представление инвариантно к сдвигу и вращению системы координат, однако оно также инвариантно к отражению системы координат. Но два зеркально отраженных белка – это не одно и то же.
Вариант 3. Углы между всеми атомами в цепочке. Такое представление тоже инвариантно к отражению системы координат. Кроме того, оно неустойчиво: вся структура сильно меняется при небольших изменениях углов.
В AlphaFold 1 нейросеть предсказывала попарные расстояния и углы между атомами в цепочке. Координаты атомов затем вычислялись на основе этих данных градиентным спуском.
В AlphaFold 2 нейросеть напрямую предсказывает координаты атомов, а также оценивает уверенность в предсказаниях. В качестве основной функции потерь используется Frame Aligned Point Error (FAPE). Эта функция инвариантна к смене системы координат как в предсказанных, так и в эталонных данных.
Более детально блок Structure module и функцию потерь FAPE мы будем рассматривать в соответствующем разделе.
Recycling
В общем виде механизм recycling может быть применен к моделям, которые способны уточнять приблизительный ответ, используя дополнительные данные, то есть к моделям следующего вида:
Инференс с recycling заключается в инициализации ответа нулями и запуске модели выбранное число () раз, используя входные данные и предыдущий ответ.
Recycling рассматривается лишь как дополнительный механизм, повышающий точность, поэтому мы хотели бы, чтобы даже после первой итерации модель выдавала бы ответ с хорошей точностью. В качестве функции потерь можно выбрать среднее значение ошибки по всем итерациям:
Если модель дифференцируема, то такую функцию потерь можно минимизировать напрямую. Однако это приведет к существенному перерасходу памяти и времени вычислений, по сравнению с однократным запуском модели. Авторы предлагают другой подход: на каждом шаге обучения выбирается случайное число между 1 и (общее для всех примеров в батче), модель запускается раз, и градиент ошибки распространяется только по последней итерации.
Подробнее см. Supplementary Material, раздел 1.10
Применение recycling в AlphaFold
AlphaFold использует в качестве входных данных шаблоны, то есть трехмерные структуры эволюционно схожих белков. Эти шаблоны играют роль гипотез о том, какой может быть структура белка. Выходными данными также является трехмерная структура, поэтому к AlphaFold может быть применен механизм recycling (рис. 10). Для этого блок input feature embeddings должен быть модифицирован таким образом, чтобы использовать не только 6 входных массивов данных, но и выходные и промежуточные данные, полученные на предыдущей итерации:
На первой итерации эти значения инициализируются нулями.
Рис. 10. Общая схема AlphaFold с recycling iterations.Авторы экспериментально подтверждают, что добавление механизма recycling в AlphaFold улучшает точность предсказаний – особенно это проявляется на тех белках, для которых таблица MSA содержит недостаточно информации.
Еще одна (несущественная) деталь заключается в том, что во время инференса evoformer stack запускается параллельно 3 раза на разных подвыборках таблицы MSA, и полученные выходные данные усредняются.
См. также Supplementary Material, Algorithm 2
Теперь начнем подробное рассмотрение каждого блока архитектуры.
Input feature embeddings
Блок Input feature embeddings (рис. 11) принимает на вход следующий набор данных:
Массивы, полученные в результате предварительной обработки данных:
– данные об аминокислотах целевого белка.
– данные о том, какую часть целевого белка мы обрезали.
– данные о кластеризованной таблице MSA.
– данные о дополнительных последовательностях в MSA.
– данные о шаблонах, в т. ч. попарные расстояния между атомами в каждом шаблоне.
– данные о шаблонах: аминокислоты и углы между атомами в каждом шаблоне.
Данные с предыдущей итерации (recycling), которые на первой итерации заменяются массивами из нулей:
– массив внутренних представлений (эмбеддингов) для каждой позиции.
– массив внутренних представлений (эмбеддингов) для каждой пары позиций.

– координаты атомов -углерода (или -углерода для глицина) в предсказанной моделью трехмерной структуре (см. также раздел «данные о шаблонах»).
Общий смысл всех выполняемых действий в том, что информация, относящаяся по смыслу к одной позиции, идет в MSA representation, а информация, относящаяся по смыслу к паре позиций, идет в pair representation. Таким образом, в MSA representation собирается вся доступная информация о позициях, а в pair representation собирается вся доступная информация о парах позиций.
Pair representation
Начнем с того, как и преобразуются в . — это последовательность векторов, соответствующих позициям в целевом белке. Каждый вектор состоит из нулей и только одной единицы (one-hot кодирование). Слой переводит каждый вектор из исходной размерности в размерность . Применяя два таких слоя, мы получаем массивы и .
Применяя два таких слоя, мы получаем массивы и .
Обработка вектора, полученного one-hot кодированием, с помощью полносвязного слоя – это то же самое, что слой Embedding в NLP-архитектурах. То есть каждой аминокислоте сопоставляется обучаемый вектор.
Чтобы получить pair representation, мы выполняем внешнюю сумму двух последовательностей векторов:
Далее с помощью массива мы должны выполнить позиционное кодирование (как в трансформере). Для этого создается обучаемый вектор-эмбеддинг для чисел (всего 65 векторов). Вектор рассчитывается следующим образом: ищется число из множества , ближайшее к , и в качестве подставляется вектор-эмбеддинг, соответствующий этому числу.
Таким образом, в позиционном кодировании стираются различия между всеми расстояниями меньше -32 и больше 32. Авторы комментируют это так:
Since we are clipping by the maximum value 32, any larger distances within the residue chain will not be distinguished by this feature.
This inductive bias de-emphasizes primary sequence distances. Compared to the more traditional approach of encoding positions in the frequency space [Vaswani et al. 2017], this relative encoding scheme empirically allows the network to be evaluated without quality degradation on much longer sequences than it was trained on.
Полученные вектора позиционного кодирования прибавляются к .
MSA representation
Для получения MSA representation мы используем и набор MSA-кластеров .
Аналогично, каждый вектор и каждый вектор обрабатываются полносвязными слоями, получая массивы и . Однако массив получается двухмерный, а массив трехмерный. Мы добавляем к массиву новую ось, повторяя его столько раз, сколько было кластеров в MSA. Затем складываем полученные массивы.
Extra MSA representation мы получаем аналогичным способом, но уже без участия .
Подробнее см. Supplementary Material, раздел 1.5
Использование информации о шаблонах
Каждый вектор в , который содержит данные о шаблонах и углах в них, обрабатывается нейронной сетью со скрытым слоем (Linear-ReLU-Linear) для получения вектора-эмбеддинга, и полученный эмбеддинг конкатенируется с MSA representation.
Каждый вектор в , обрабатывается полносвязным слоем для получения эмбеддинга (рис. 8, слой «embed»). Далее этот эмбеддинг обрабатывается цепочкой операций, которые не отражены на рис. 8, а именно:
Triangular self-attention around starting node
Triangular self-attention around ending node
Triangular multiplicative update using outgoing edges
Triangular multiplicative update using incoming edges
Pair transition
Эти же операции применяются в evoformer-блоках, поэтому мы будем разбирать их позднее, при описании evoformer-блока. После применения этих операций мы получаем массив размером (, , , 128) – эмбеддинг каждой пары позиций в каждом шаблоне.
Затем мы добавляем эту информацию к pair representation, используя механизм template pointwise attention. Это вариант multi-head self-attention (Vaswani et al. 2017), в котором запросом (query) является -й элемент pair representation, а ключами и значениями — -е элементы каждого из шаблонов. Более детально останавливаться на этой операции сейчас не будем.
Более детально останавливаться на этой операции сейчас не будем.
Подробнее см. Supplementary Material, раздел 1.7.1
Также в блоках «R» (recycling) мы используем информацию с предыдущей итерации. Для экономии места также не будем разбирать принцип работы блока «R», поскольку он является лишь дополнительным инструментом.
Подробнее см. Supplementary Material, раздел 1.10
Evoformer
Блок evoformer принимает и возвращает два массива:
MSA representation, размером – эмбеддинги пар (позиция + последовательность).
Pair representation, размером – эмбеддинги пар (позиция + позиция)
И устроен из блоков, имеющих собственные обучаемые веса (рис. 12).
Рис. 12. Схема вычислений в evoformer-блоке.Evoformer похож на трансформер, поскольку многие операции в нем используют self-attention. Как в и трансформере, через все операции проброшены skip connections, то есть входные данные прибавляются к выходным. Операция, вокруг которой проброшен skip connection, называется residual-блоком. В некоторых residual-блоках добавляется dropout по отдельным столбцам и строкам. Общий алгоритм вычислений в evoformer stack, дублирующий рис. 12, приведен в алгоритме 6.
Операция, вокруг которой проброшен skip connection, называется residual-блоком. В некоторых residual-блоках добавляется dropout по отдельным столбцам и строкам. Общий алгоритм вычислений в evoformer stack, дублирующий рис. 12, приведен в алгоритме 6.
Большая часть операций, выполняемых в evoformer-блоке, основаны на criss-cross attention. Этот подход заключается в следующем: имея трехмерный массив, каждый -й элемент которого является вектором-эмбеддингом, мы сначала применяем операцию multi-head self-attention к каждой строке массива, затем к каждому столбцу (или наоборот).
Надо так же отметить, что evoformer-блоки, обрабатывающие MSA-кластеры, и evoformer-блоки, обрабатывающие дополнительные MSA-последовательности (см. рис. 9, extra evoformer stack), имеют некоторые отличия. Эти отличия обусловлены тем, что дополнительные таблицы MSA содержат большое количество последовательностей (в отличие от MSA-кластеров), и обрабатывающие их evoformer-блоки должны быть адаптированы для работы с большим количеством последовательностей.
Подробнее см. Supplementary Material, раздел 1.7.2
Далее мы последовательно разберем все вычислительные элементы evoformer-блока (рис. 12).
MSA row-wise and column-wise gated self-attention
Вспомним, что массив MSA representation состоит из набора последовательностей, каждая из которых состоит из набора позиций. Каждая позиция представлена вектором-эмбеддингом длиной 256. Таким образом, массив MSA representation имеет три оси, и его можно рассматривать как таблицу из векторов-эмбеддингов.
Первые два элемента evoformer-блока (row-wise self-attention, column-wise self-attention) отвечают за обновление MSA representation, при котором в векторы-эмбеддинги «обмениваются информацией» друг с другом (рис. 13, 14). Кроме того, в row-wise self-attention используется информация из pair representation.
Если в обеих схемах убрать gating, а в первой также убрать pair bias, то мы получим в точности механизм multi-head self-attention, используемый в трансформерах. Этот механизм я подробно разбирал в обзоре статьи о трансформерах (см. разделы с «Dot-product attention» до «Multi-head attention»).
Этот механизм я подробно разбирал в обзоре статьи о трансформерах (см. разделы с «Dot-product attention» до «Multi-head attention»).
В row-wise self-attention обмен информацией между векторами идет в пределах одной строки (то есть между всеми позициями в белке), в column-wise self-attention – в пределах одного столбца (то есть между одной и той же позицией во всех последовательностях).
Рис. 13. MSA row-wise gated self-attention with pair bias.Рис. 14. MSA column-wise gated self-attention.Gating заключается в том, что взвешенные средние векторов умножаются на «маску», полученную с помощью сигмоиды (рис. 13, 14). Смысл добавления gating в работе не объясняется, но это похоже на механизм multiplicative input gate и forget gate в LSTM.
Pair bias заключается в добавлении к скалярным произведениям векторов дополнительного слагаемого, рассчитанного как линейное преобразование каждого вектора в pair representation. Таким образом нейронная сеть может научиться учитывать информацию из pair representation при обновлении MSA representation. Если удалить pair bias, то pair representation никак не будет влиять на MSA representation в evoformer-блоке, что противоречит идее о том, что эти массивы должны меняться под действием друг друга.
Если удалить pair bias, то pair representation никак не будет влиять на MSA representation в evoformer-блоке, что противоречит идее о том, что эти массивы должны меняться под действием друг друга.
В качестве альтернативы, можно было бы заменить row-wise и column-wise attention на общий attention между всеми возможными парами векторов в MSA representation. Однако такой способ привел бы к намного более тяжелым вычислениям и большему расходу памяти. Используя разложение attention на row-wise и column-wise, авторы ссылаются на работу CCNet: Criss-Cross Attention for Semantic Segmentation (2018).
Стоит упомянуть и еще одну деталь, не отраженную на рис. 13, 14. Каждый входной вектор в MSA representation и pair representation нормализуется L2-нормализацей до единичной длины (Layer Normalization). Алгоритм 7 дублирует рис. 13. В этом алгоритме MSA representation обозначается как , pair representation обозначается как .
Алгоритм column-wise self-attention устроен аналогично, но без использования pair representation.
MSA transition
Следующий элемент evoformer-блока – MSA transition, в котором каждый вектор в MSA-таблице преобразуется с помощью нейронной сети с одним скрытым слоем (рис. 15).
Рис. 15. MSA transition.Здесь снова все устроено так же, как в блоке трансформера. Как и в трансформере, вокруг MSA transition также проброшена связь skip connection (см. рис. 12). Также отметим, что перед первым слоем Linear выполняется операция LayerNormalization (не отражена на схеме).
Важно, что эта операция выполняется независимо и одинаково по каждому вектору в таблице MSA. Обозначим сеть со скрытым полносвязным слоем за . Тогда для любых , верно следующее: (используя numpy-индексацию).
Outer product mean
Блок outer product mean (рис. 16) обновляет pair representation под действием MSA representation.
Обозначим pair representation символом (), MSA representation символом (). Допустим, мы хотим обновить вектор . В таблице MSA representation индексам и соответствуют два столбца: и . Эти столбцы нужно использовать для обновления . Фактически, нам достаточно лишь придумать способ передачи информации из MSA в . Какая конкретно информация будет передаваться – нейронная сеть определит сама в ходе обучения.
В таблице MSA representation индексам и соответствуют два столбца: и . Эти столбцы нужно использовать для обновления . Фактически, нам достаточно лишь придумать способ передачи информации из MSA в . Какая конкретно информация будет передаваться – нейронная сеть определит сама в ходе обучения.
Авторы предлагают поступать следующим образом (рис. 13). Каждый вектор-эмбеддинг столбцов и обрабатывается полносвязным слоем (для уменьшения размерности векторов-эмбеддингов с 256 до 32), и мы получаем две последовательности векторов.
Далее считаем внешнее произведение (outer product). Разберем эту операцию детально. Обе входные последовательности ( и ) имеют размер (, 32), где — количество кластеров в MSA. Результат имеет размер (, 32, 32). Для каждого , , : .
Рис. 16. Outer product mean.Полученный массив с тремя осями сначала усредняется по оси, соответствующей номеру кластера, затем «вытягивается» в вектор и обрабатывается полносвязным слоем. В результате получаем вектор длиной 128, который прибавляется к вектору .
Назовем каждый элемент вектора-эмбеддинга «признаком». Тогда суперпозиция внешнего произведения и усреднения означает, что мы считаем все попарные скалярные произведения -го признака и -го признака , то есть матрицу Грама. Это напоминает подсчет матрицы Грама между признаками в сверточных сетях при переносе стиля (см. Image Style Transfer Using Convolutional Neural Networks).
Авторы добавляют, что операция outer product mean является затратной по памяти, так как в ходе нее рассчитываются промежуточные тензоры большой размерности.
Triangular multiplicative update
Если бы массив pair representation состоял из попарных расстояний между вершинами, тогда важным было бы соблюдение неравенства треугольника: для любых расстояние между позициями и должно быть не больше, чем сумма расстояния между позициями и и расстояния между позициями и . В противном случае построить трехмерную структуру по матрице не получится.
Не исключено, что авторы в ходе работы рассматривали и такой вариант, но решили отказаться от него в пользу более репрезентативного, когда каждая пара позиций кодируется не одним числом, а вектором-эмбеддингом.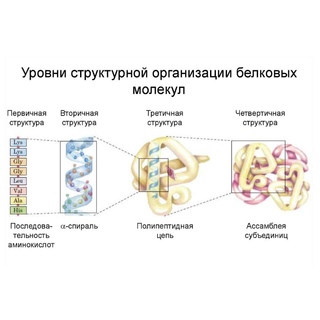 В этом случае сеть должна сама обучиться неравенству треугольника на эмбеддингах.
В этом случае сеть должна сама обучиться неравенству треугольника на эмбеддингах.
Элемент pair representation является вектором-эмбеддингом пары позиций , или, иначе говоря, эмбеддингом ориентированного ребра . Механизм triangular multiplicative update является обучаемым преобразованием, которое работает по очереди со всеми возможными тройками ребер в pair representation: для всех обновляет эмбеддинг -го ребра с помощью эмбеддингов ребер и , а также с помощью эмбеддингов ребер и (рис. 17). Поэтому triangular multiplicative update присутствует в evoformer-блоке в двух экземплярах: «outgoing edges» и «incoming edges».
Рис. 17. Triangular multiplicative update.На рис. 18 и в алгоритме 11 (см. ниже) показана вычислительно эффективная матричная форма triangular multiplicative update. Операции, выполняемые в строке 4 алгоритма, линейны, поэтому знак суммы можно вынести, и таким образом Algorithm 11 можно рассмотреть как последовательность выполнений Algorithm 11 для каждого фиксированного . Благодаря этому можно упростить понимание операции triangular multiplicative update: ее можно рассмотреть как операцию, выполняемую по очереди для каждой тройки .
Благодаря этому можно упростить понимание операции triangular multiplicative update: ее можно рассмотреть как операцию, выполняемую по очереди для каждой тройки .
Назовем ребро целевым ребром, а ребра и смежными ребрами. Вектор-эмбеддинг целевого ребра обрабатывается полносвязным слоем с сигмоидой («5» на рис. 15), полученный вектор назовем , вектора-эмбеддинги смежных ребер обрабатываются операцией , получая пару векторов и . Полученная пара векторов поэлементно умножается («6» на рис. 14), нормализуется L2-нормализацией до единичной длины (LayerNormalization) и снова обрабатывается полносвязным слоем, полученный вектор назовем . Далее вектора и поэлементно умножаются, и результат записывается в выходной массив на место эмбеддинга ребра .
Резюмируя: для всех троек чисел к ребру добавляется слагаемое, являющееся функцией от ребер , , . Под «ребром» понимаем эмбеддинг ребра в массиве pair representation.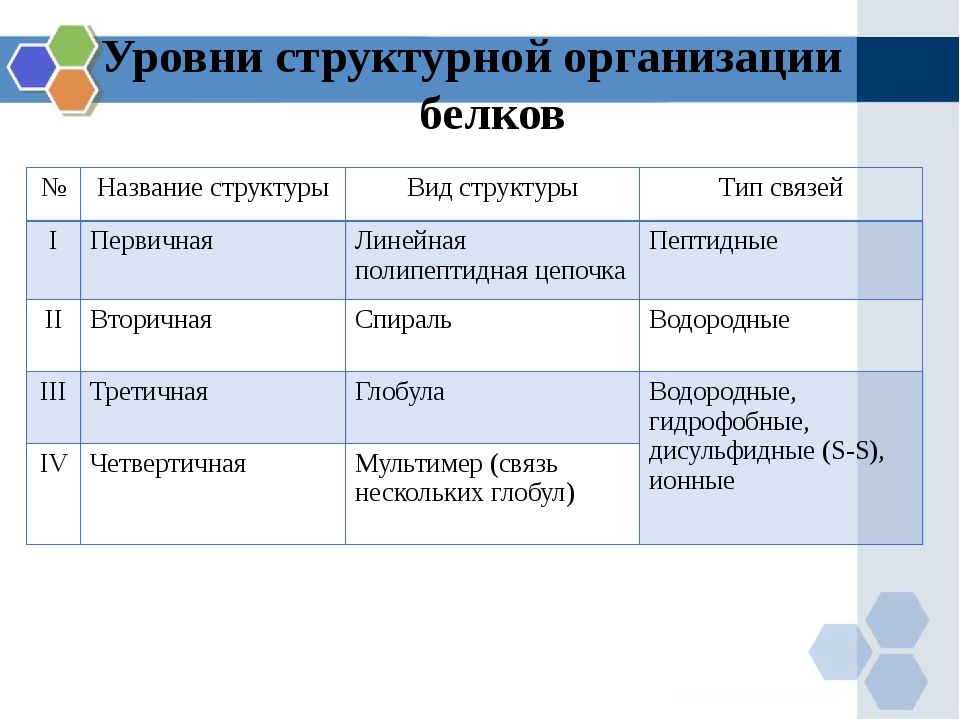
Аналогично операция повторяется для «incoming edges», только на этот раз мы берем функцию от ребер , , .
Подробнее см. Supplementary Material, раздел 1.6.5
Triangular self-attention
В операции «triangular self-attention around starting node» все ребра, исходящие из одной и той же вершины (строка в массиве pair representation), обмениваются между собой информацией. Для этого к строке массива pair representation применяется операция multi-head self-attention.
Аналогично, в операции «triangular self-attention around ending node» обмениваются информацией все ребра, входящие в одну и ту же вершину. Для этого к столбцу массива pair representation применяется операция multi-head self-attention.
Есть два отличия triangular self-attention (рис. 19) от стандартного multi-head self-attention. Первое отличие заключается в использовании gating («1» на рис. 19), так же как в MSA gated self-attention.
Рис. 19. Triangular self-attention around starting node.
Второе отличие заключается в использовании ребер и при обмене информацией между ребрами и (рис. 20). Ниже рассмотрим этот механизм более детально.
Рис. 20. Triangular self-attention.В self-attention (см. Attention Is All You Need) для обновления ребра рассчитываются скалярные произведения между query для этого ребра и keys для ребер (для всех ). Полученный набор чисел называется attention logits, или dot-product affinities (см. рис. 19). К этому набору чисел применяется softmax для получения весов. Веса затем используются для расчета взвешенного среднего.
Особенность triangular self-attention в том, что к dot-product affinities прибавляется дополнительное слагаемое («2» на рис. 19, строка 5 алгоритма 13). Например, пусть мы хотим обновить вектор-эмбеддинг ребра , используя ребро . Мы рассчитываем dot-product affinity скалярным произведением query ребра и key ребра . К полученному числу мы прибавляем еще одно число, полученное из вектора-эмбеддинга ребра с помощью линейного слоя с одним нейроном.
На рис. 19 в нижней ветке указано количество выходных нейронов , но это потому, что используется multi-head self-attention с голов. Все головы работают независимо друг от друга . В итоге операция triangular self-attention напоминает triangular multiplicative update, поскольку в ней тоже используются тройки ребер. Но отличие в том, что в triangular multiplicative update не используется механизм внимания, поэтому обмен информацией между тройкой ребер происходит без участия других ребер.
Transition in the pair stack
Эта последняя операция в evoformer-блоке выполняется аналогично операции MSA transition (рис. 15). Массив pair representation состоит из векторов-эмбеддингов каждого направленного ребра, и каждый эмбеддинг в нем обновляется с помощью полносвязной нейронной сети с одним скрытым слоем.
Structure module
Блок structure module принимает на вход данные, полученные на выходе из evoformer stack:
single representation размером – вектора-эмбеддинги для каждой позиции
pair representation размером – вектора-эмбеддинги для каждой пары позиций
Формальное представление 3D-структуры
Каждая позиция в белке – это атом -углерода, крепящийся к нему аминокислотный остаток, а также пептидная связь между атомами -углерода (см. рис. 6). В более простом варианте задачи достаточно предсказать координаты атомов -углерода («backbone»), в более сложном варианте задачи нужно предсказать координаты всех атомов белка.
рис. 6). В более простом варианте задачи достаточно предсказать координаты атомов -углерода («backbone»), в более сложном варианте задачи нужно предсказать координаты всех атомов белка.
В AlphaFold для каждой позиции в белке вводится локальная ортонормированная система координат (backbone frame). Ноль в этой системе соответствует координате атома -углерода. Координаты всех атомов аминокислотного остатка, а также атомов пептидной связи, можно описывать как в локальной системе координат, так и в глобальной.
Переход из локальной системы координат в глобальную можно описать как поворот + смещение. Поворот описывается ортогональной матрицей, смещение – вектором. Поэтому каждой позиции в белке сопоставляется 12 чисел: матрица поворота 3×3 и вектор смещения.
Авторы вводят символ для обозначения локальной системы координат -й позиции:
Переход из локальной в глобальную систему координат осуществляется умножением на матрицу и прибавлением вектора . Упорядоченную пару («фрейм») можно рассмотреть как операцию перехода между системами координат, и записывать следующим образом:
Упорядоченную пару («фрейм») можно рассмотреть как операцию перехода между системами координат, и записывать следующим образом:
Введем операцию суперпозиции двух систем координат:
В AlphaFold на первом шаге координаты всех атомов -углерода инициализируются нулями, а все матрицы – матрицами идентичности. Авторы называют этот способ «black hole initialization». Затем выполняется 8 итераций, на каждом из которых каждый фрейм обновляется путем суперпозиции с другим ортонормированным фреймом, рассчитанным нейросетью. Тем самым координаты всех атомов -углерода и их ориентации уточняются.
Описанный принцип авторы называют «residue gas»: фрейм каждой позиции находится в пространстве «сам по себе»: не задается явного ограничения, что -й и -й фрейм должны находиться друг от друга на требуемом расстоянии в глобальной системе координат. Сеть сама «выучивает» это правило в ходе обучения.
С другой стороны, при таком подходе погрешность предсказания приведет к тому, что в результате мы получим невозможную структуру, где атомы -углерода находятся друг от друга немного не на тех расстояниях, на каких должны быть. Поэтому для получения финального предсказания ответ нейросети уточняется итеративными алгоритмами минимизации энергии.
Поэтому для получения финального предсказания ответ нейросети уточняется итеративными алгоритмами минимизации энергии.
Помимо координат атомов -углерода, нужно также определить координаты всех остальных атомов. Каждый аминокислотный остаток тоже не является «жесткой» структурой. Для расчета координат атомов в аминокислотном остатке для каждой позиции в белке вводится набор углов кручения (торсионные углы): . Эти углы тоже рассчитываются в structure module. Зная фреймы и углы кручения для всех позиций, можно рассчитать координаты всех атомов в белке.
Подробнее см. Supplementary Material, раздел 1.8
На рис. 21 показано, как в AlphaFold моделируется трехмерная структура белка. Фреймы задают позиции атомов -углерода и ориентацию соседних атомов, а углы кручения позволяют определить позиции атомов в аминокислотных остатках.
Рис. 21. Residue gas.Схема вычислений в structure module
Structure module (рис. 22) состоит из 8 последовательно соединенных блоков с общими весами, то есть работает как рекуррентная сеть.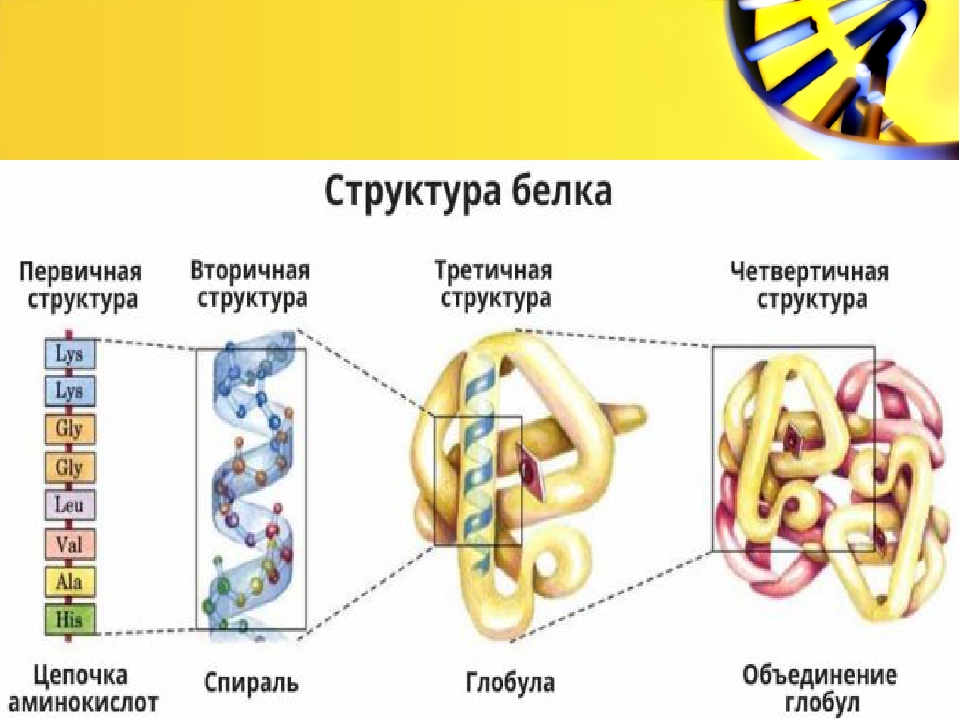 Между блоками передаются два массива данных:
Между блоками передаются два массива данных:
single representation (массив векторов-эмбеддингов), полученный из evoformer-стека
backbone frames (фреймы каждой позиции), которые изначально инициализируются методом «black hole initialization», описанным выше.
Рассмотрим операции, выполняемые внутри каждого блока.
Invariant point attention. В ходе этой операции массив single representation обновляется под действием себя самого (self-attention), а также под действием pair representation и backbone frames. Вокруг invariant point attention проброшена связь skip connection.
Transition. Каждый вектор-эмбеддинг в single representation обновляется полносвязной нейронной сетью (по аналогии с тем, как в трансформере полносвязная нейронная сеть применяется после self-attention).
Backbone update. Backbone frames обновляются под действием single representation.
Predict angles. Углы кручения рассчитываются с помощью текущего single representation и single representation, полученного из evoformer-стека.
(координаты атомов -углерода) извлекаются из backbone frames, для этого берется второй компонент каждого фрейма.
В результате, выходными данными каждого блока (intermediate predictions) являются координаты атомов -углерода и углы кручения для каждого аминокислотного остатка. Выходные данные последнего блока являются финальным предсказанием. По углам кручения рассчитываются координаты всех атомов в белке (all atom coordinates). Также рассчитывается уверенность в предсказаниях (confidence).
Авторы отмечают, что для стабилизации обучения AlphaFold блокируют «протекание» градиента по матрицам поворота из следующего блока в предыдущий. Технически это означает следующее: в том месте, где фреймы передаются из одного блока в другой, добавляется операция StopGradient – это тождественное преобразование, градиент которого переопределяется значением 0.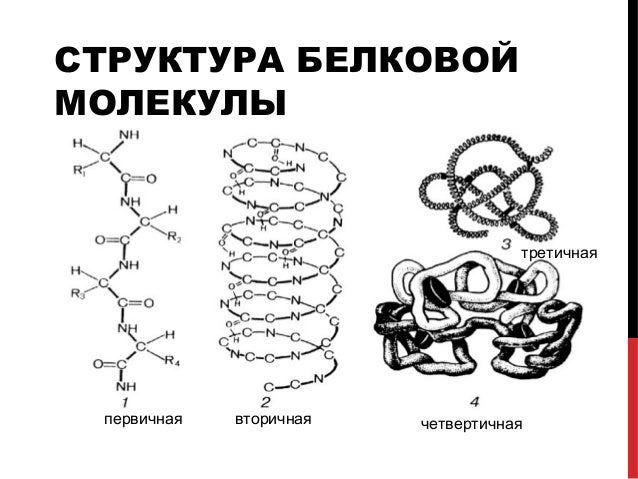
We found it helpful to zero the gradients into the orientation component of the rigid bodies between iterations (Algorithm 20 line 20), so any iteration is optimized to find an optimal orientation for the structure in the current iteration, but is not concerned by having an orientation more suitable for the next iteration. Empirically, this improves the stability of training, presumably by removing the lever effects arising in a chained composition frames.
Далее более подробно рассмотрим выполняемые вычисления и функции потерь.
Invariant point attention
Схема вычислений в блоке invariant point attention показана на рис. 23. Красным цветом показана стандартная операция multi-head self-attention, применяемая к single representation. Синим цветом показано использование pair representation в расчете attention logits, а также при обновлении single representation. Использование backbone frames (зеленый цвет) подробнее разберем далее.
Операция invariant point attention спроектирована так, что является инвариантной к смене глобальной системы координат в backbone frames. Это важное свойство: обновление single representation должно происходить с учетом структуры белка (точнее, ее приближения на текущей итерации), которая не зависит от выбранной системы координат.
В алгоритме 22 показано, как конкретно используются backbone frames. Для упрощения можно принять и избавиться от индексов в алгоритме. Зафиксируем пару позиций и . Этим позициям соответствуют вектора-эмбеддинги и фреймы .
Линейными преобразованиями рассчитываются 4 вектора в трехмерном пространстве: (query points). Эти вектора переводятся из локальной системы координат в глобальную систему координат.
Линейными преобразованиями рассчитываются 4 вектора в трехмерном пространстве: (key points). Эти вектора переводятся из локальной системы координат в глобальную систему координат.
В результате получаем 4 вектора-ключа и 4 вектора значения в глобальной системе координат. Между парами этих векторов рассчитываются квадраты расстояний, и складываются. Результат умножается на константу и добавляется к dot-product affinities (строка 7 в алгоритме 22). Данная операция является инвариантной к смене глобальной системы координат, что ясно из геометрических соображений.
Смысл данной операции, по-видимому, следующий: обучившись использовать подходящие вектора queries и keys, в зависимости от типа аминокислотного остатка, нейронная сеть может научиться моделировать взаимодействия между разными аминокислотными остатками. Полученные вектора в локальной системе координат могут означать некие ключевые точки для данного остатка. Расстояния между парами точек могут кодировать взаимодействие двух аминокислотных остатков.
Теперь рассмотрим, как backbone frames используются в строке 11 алгоритма 22, где на основании attention weights обновляется вектор single representation.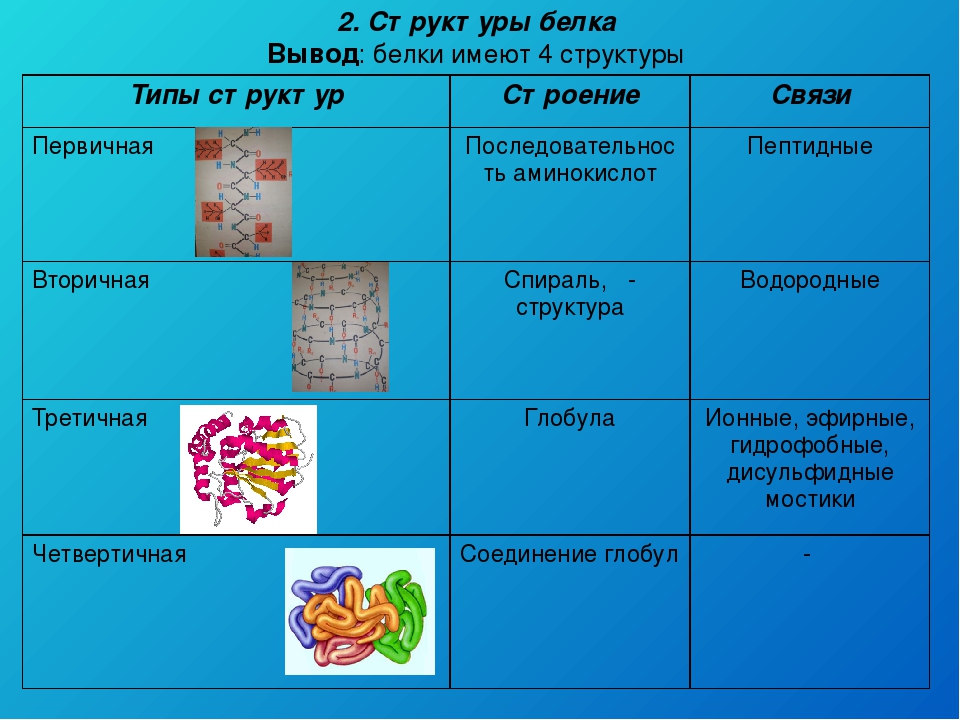
Обновляя -ю позицию, мы имеем attention weights для каждой -й позиции . Для каждой -й позиции рассчитывается 8 векторов в локальной системе координат : (point values). Эти вектора переводятся в глобальную систему координат, где считается из взвешенное среднее с помощью весов . Полученный вектор переводится в локальную систему координат , и результат после линейного преобразования добавляется к эмбеддингу -го вектора. Такая операция напоминает расчет «взвешенного центра масс» и тоже является инвариантной к преобразованиям глобальной системы координат.
Доказательство инвариантности см. в Supplementary Material, раздел 1.8.2
Другие операции в structure module
Операция transition (см. рис. 22) является нейронной сетью с 2 скрытыми слоями, вокруг которой проброшена связь skip connection. Также в начале и конце добавляются LayerNorm и Dropout.
Операция backbone update заключается в коррекции каждого фрейма с помощью матрицы поворота и вектора смещения.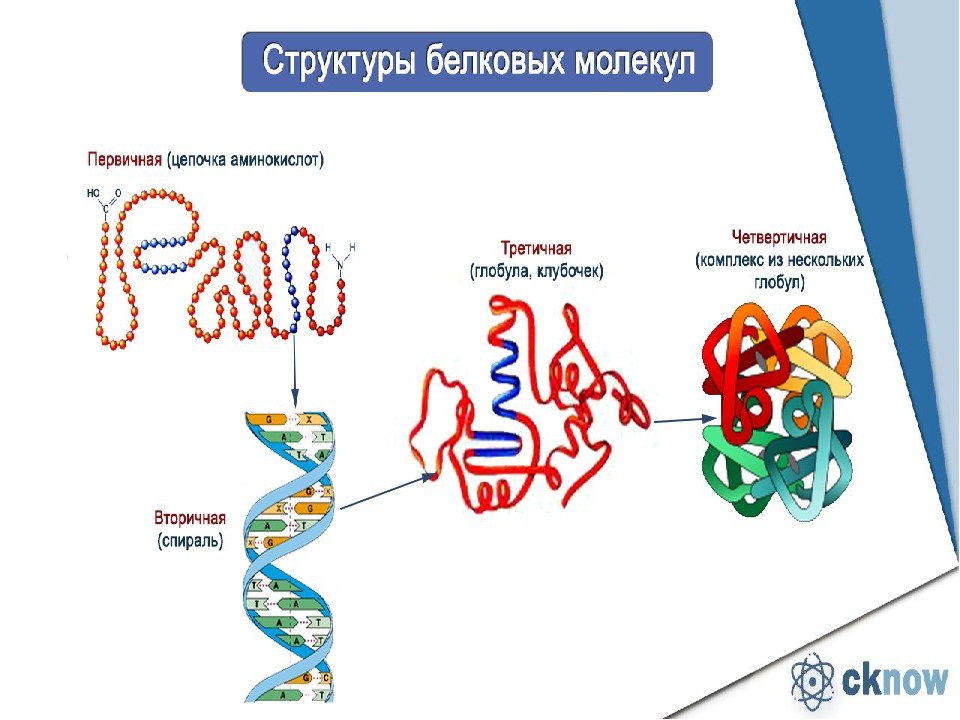 Любой поворот в трехмерном пространстве можно описать кватернионом, первый элемент которого равен 1. Кватернион рассчитывается линейным преобразованием эмбеддинга -й позиции, и затем преобразуется в матрицу поворота (алгоритм 23).
Любой поворот в трехмерном пространстве можно описать кватернионом, первый элемент которого равен 1. Кватернион рассчитывается линейным преобразованием эмбеддинга -й позиции, и затем преобразуется в матрицу поворота (алгоритм 23).
Подробнее см. Supplementary Material, раздел 1.8.3
Операция predict angles позволяет получить углы кручения для -й позиции с помощью эмбеддинга этой позиции. Для этого используется нейронная сеть с несколькими слоями и skip connections, которая также использует изначальный эмбеддинг -й позиции, полученный из evoformer-блока. Углы предсказываются как точки на единичной окружности: рассчитывается вектор из двух чисел и нормализуется до единичной длины.
Подробнее см. Supplementary Material, Algorithm 20, строки 11-14
Влияние архитектуры structure module на метрику качества
Несмотря на сложность операций, выполняемых в structure module, их положительное влияние оказывается лишь незначительным. Авторы демонстрируют, что радикальное упрощение structure module, включая избавление от рекуррентности и invariant point attention, а также отказ от использования pair representation в structure module ведет лишь к незначительному ухудшению метрики качества.
Авторы демонстрируют, что радикальное упрощение structure module, включая избавление от рекуррентности и invariant point attention, а также отказ от использования pair representation в structure module ведет лишь к незначительному ухудшению метрики качества.
См. также Supplementary Material, раздел 1.13, Figure 10, “No IPA”
Функции потерь в AlphaFold
Как это часто бывает в сложных архитектурах, в AlphaFold минимизируется сумма нескольких разных функций потерь. Обучение AlphaFold проходит в два этапа. Второй этап, называемый fine-tuning, отличается большим размером кропа и таблицы MSA, меньшим learning rate, а также в нем добавлены еще две функции потерь (рис. 24).
Рис. 24. Функции потерь в AlphaFold. – основная функция потерь в AlphaFold, сравнивающая предсказанную 3D-структуру белка с эталонной. Она рассчитывается на последней итерации в structure module (final loss на рис. 22). Далее эта функция потерь будет рассмотрена подробнее.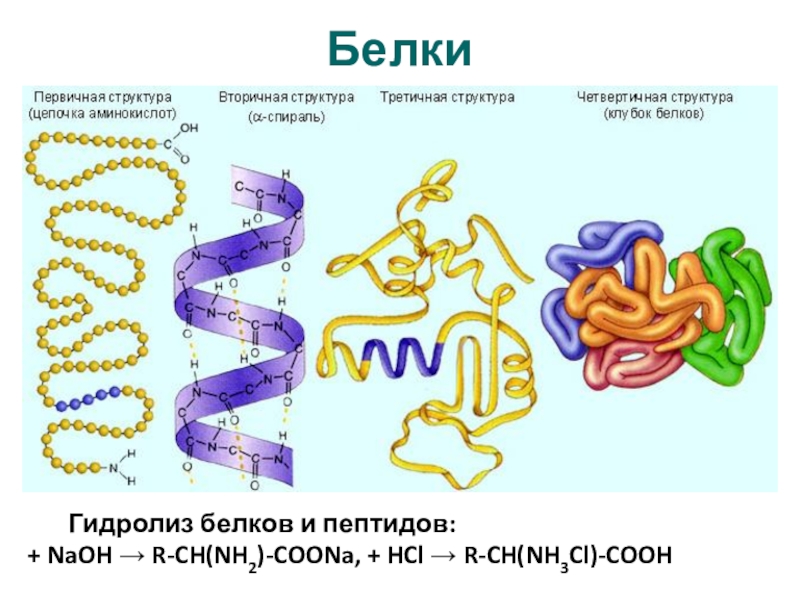
– дополнительная функция потерь (auxiliary loss), рассчитываемая после каждой итерации в structure module. Эта функция является суммой двух слагаемых. Первое слагаемое – упрощенный вариант FAPE, в котором рассчитывается ошибка предсказания только для атомов -углерода (а не для всех атомов белка). Второе слагаемое – torsion angle loss, который сравнивает предсказанные углы кручения с эталонными (подробнее см. далее).
Torsion angle loss рассматривается как дополнительная функцией потерь, поскольку минимизация основной функции потерь FAPE должна приводить также и к минимизации ошибки предсказания углов. Но добавление дополнительных функций потерь в тех местах, где мы знаем, как должен выглядеть ответ, может улучшить стабильность и качество обучения.
The purpose of the FAPE, aux, distogram, and MSA losses is to attach an individual loss to each major subcomponent of the model (including both the pair and MSA final embeddings) as a guide during the training of the “purpose” of each unit.

Подробнее см. Supplementary Material, раздел 1.9
Чтобы уменьшить относительную важность коротких последовательностей, суммарная функция потерь умножается на квадратный корень из длины белка (которая ограничена сверху размером кропа). Такое действие побуждает модель повышать точность предсказания на длинных белках, особенно учитывая тот факт, что их меньше, чем коротких (см. рис. 5).
Далее рассмотрим подробнее FAPE и torsion angle loss, затем остальные функции потерь.
Frame aligned point error (FAPE)
FAPE – основная функция потерь AlphaFold, которая сравнивает предсказанную 3D-структуру с эталонной. Для понимания функции FAPE понадобится снова вспомнить понятие «фрейм» (backbone frame) – локальная ортонормированная система координат, связанная с -м атомом -углерода. Подробнее о понятии фрейма см. раздел «Формальное представление 3D-структуры».
Функция FAPE может быть реализована в двух вариантах: либо она принимает только координаты атомов -углерода, либо координаты всех атомов белка. Рассмотрим только первый случай как более простой. В этом случае в алгоритме 28 является началом координат во фрейме , и аналогично является началом координат во фрейме . Тогда второй и четвертый аргументы излишни, и можно считать, что функция принимает только два аргумента:
Рассмотрим только первый случай как более простой. В этом случае в алгоритме 28 является началом координат во фрейме , и аналогично является началом координат во фрейме . Тогда второй и четвертый аргументы излишни, и можно считать, что функция принимает только два аргумента:
Зафиксировав пару позиций и , можно рассчитать координату -го атома во фрейме, связанном с -м атомом. Выполним такое действие в предсказанных и в эталонных координатах, мы получим векторы и , которые не будут зависеть от поворота и смещения глобальной системы координат. Далее считаем расстояние между и , которое ограничиваем сверху значением 10 ангстрем. Полученные значения усредняем по всем .
Функция FAPE является инвариантной к повороту и смещению глобальной системы координат как в предсказанном, так и в эталонном наборе координат, но не инвариантна к отражению системы координат, что важно при предсказании структуры белков.
В AlphaFold 2 функция потерь FAPE для атомов -углерода рассчитывается после каждой итерации в structure module (auxiliary losses на рис. 22). Плюс к этому, на последней итерации рассчитывается FAPE для всех атомов в белке (final loss на рис. 22).
22). Плюс к этому, на последней итерации рассчитывается FAPE для всех атомов в белке (final loss на рис. 22).
Еще одна особенность заключается в том, что некоторые аминокислотные остатки симметричны. Например, симметричным является остаток тирозина. Если в предсказанной структуре остаток будет повернут на 180 градусов относительно эталонной структуры, то ответ будет тоже правильным, хотя координаты отдельных атомов в FAPE не совпадут. Чтобы решить эту проблему, авторы вводят дополнительную операцию «rename symmetric ground truth atoms», которая делает предсказание по возможности более похожим на ответ, «разворачивая» симметричные остатки на 180 градусов.
Подробнее см. Supplementary Material, разделы 1.9.2-1.9.5, 1.8.5
Torsion angle loss
Углы кручения в AlphaFold предсказываются как точки на единичной окружности. Как было описано выше, для предсказания углов в AlphaFold сначала рассчитывается произвольный вектор из двух чисел, а затем этот вектор нормализуется до единичной длины. На каждой итерации в structure module рассчитываются углы (см. рис. 22), и добавляются две дополнительные функции потерь:
На каждой итерации в structure module рассчитываются углы (см. рис. 22), и добавляются две дополнительные функции потерь:
Первая функция потерь «штрафует» слишком большие или слишком маленькие вектора до нормализации, чтобы предотвратить стремление этих векторов к нулю или бесконечности.
Вторая функция потерь сравнивает предсказанные углы с эталонными, рассчитывая L2-норму разности между ними. Однако некоторые аминокислотные остатки симметричны, и угол 180° эквивалентен углу 0°. Это учитывается путем предоставления «альтернативных» эталонных углов.
Подробнее см. Supplementary Material, раздел 1.9.1
MSA loss
– точность предсказания ячеек таблицы MSA, закрытых маской. На этапе подготовки данных маскировалось 15% ячеек таблицы MSA (см. раздел «Кластеризация и маскирование таблицы MSA»). Задача их предсказания аналогична задаче «masked language model» в языковой модели BERT. Для предсказания используется массив MSA representations, полученный на выходе из evoformer stack. По каждому вектору в MSA representations линейным слоем осуществляется классификация в один из типов аминокислотных остатков, и в качестве функции потерь используется кроссэнтропия.
По каждому вектору в MSA representations линейным слоем осуществляется классификация в один из типов аминокислотных остатков, и в качестве функции потерь используется кроссэнтропия.
Подробнее см. Supplementary Material, раздел 1.9.9
Distogram loss
– loss предсказания дистограммы. Дистограммой называется матрица попарных расстояний между атомами -углерода. Для предсказания дистограммы авторы добавляют в модель дополнительный выходной слой (distogram head), который линейно отображает каждый вектор из pair representation в распределение вероятностей для элемента дистограммы. Пространство делится на 64 интервала, и задача рассматривается как классификация с 64 классами, каждый класс соответствует одному из интервалов – то есть здесь используется тот же подход, что и при создании дистограмм шаблонов (см. раздел «Данные о шаблонах»).
Подробнее см. Supplementary Material, раздел 1.9.8
Авторы проводят дополнительные эксперименты, удаляя или упрощая различные блоки в AlphaFold и изучая, как это скажется на качестве. Выясняется, что во-первых отказ от использования distogram loss ведет лишь к незначительному падению качества предсказаний.
Выясняется, что во-первых отказ от использования distogram loss ведет лишь к незначительному падению качества предсказаний.
Во-вторых, блокировка протекания градиента из structure module в evoformer stack (то есть теперь evoformer stack обучается только под действием distogram loss и MSA loss) ведет к существенному падению качества. Это говорит о том, что минимизировать лишь distogram loss недостаточно для качественного обучения.
Вспомним, что радикальное упрощение structure module вело лишь к небольшому падению точности. Это говорит о том, что для высокой точности предсказаний важна не столько сложность structure module, сколько его наличие, то есть предсказание 3D-структуры, а не дистограммы. Возможно это связано с тем, что дистограмме соответствует две зеркальные структуры, и лишь одна из них правильная.
См. также Supplementary Material, раздел 1.13, Figure 10
Confidence loss
– loss для оценки уверенности в предсказаниях. Уверенность оценивается следующим образом: между предсказанной и эталонной структурой для каждой позиции рассчитывается метрика локального сходства LDDT (специфичная для задачи предсказания структуры белка), а затем модель учится предсказывать рассчитанное значение LDDT для каждой позиции.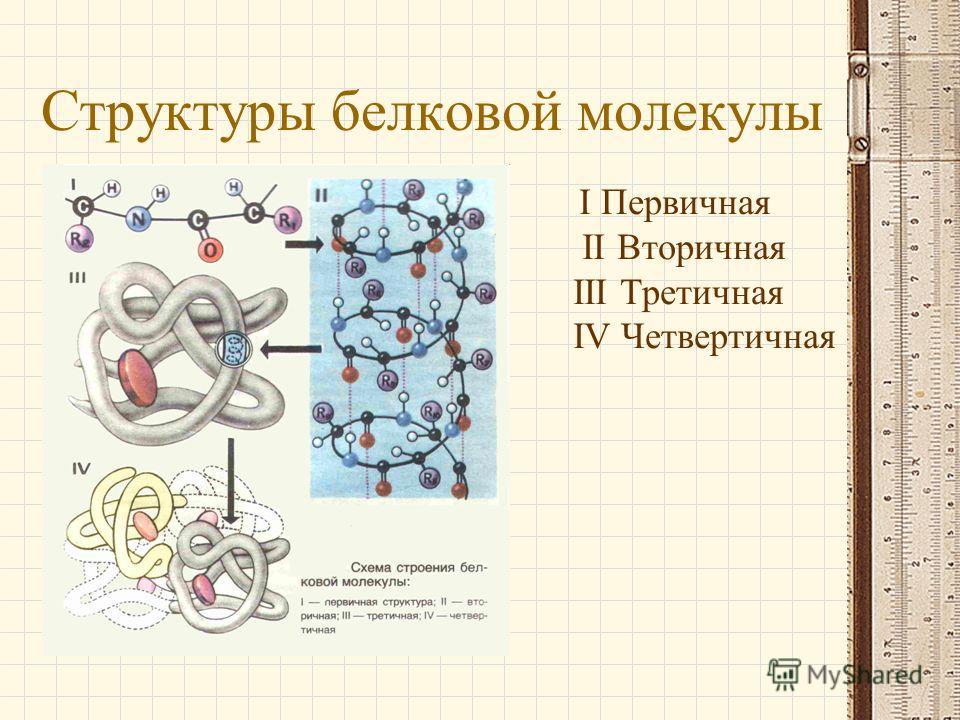 На рис. 25 показана предсказанная структура белка (другие структуры можно посмотреть здесь).
На рис. 25 показана предсказанная структура белка (другие структуры можно посмотреть здесь).
Violation losses
– сумма функций потерь, которые «штрафуют» предсказанные структуры, невозможные физически. Такое иногда происходит из-за применения концепции «residue gas» (см. раздел «Формальное представление 3D-структуры»). Violation losses рассчитываются только на втором этапе обучения (fine-tuning), тем самым модель подталкивается к тому, чтобы предсказывать физически корректные структуры даже в тех случаях, когда она не уверена в предсказаниях.
“Violation” losses encourage the model to produce a physically plausible structure with correct bond geometry and avoidance of clashes, even in cases where the model is highly unsure of the structure. … Using the violation losses early in training causes a small drop in final accuracy since the model overly optimizes for the avoidance of clashes, so we only use this during fine-tuning.
Процесс обучения AlphaFold
Обучение AlphaFold выполняется в два этапа, как было описано в разделе «Функции потерь в AlphaFold». Также при обучении используется механизм recycling, как было описано в разделе «Применение recycling в AlphaFold».
Исходный код можно найти в этом репозитории. Для подготовки данных использовался пайплайн TensorFlow версии 1.x, для прямого и обратного прохода использовалась библиотека JAX.
Оптимизатор и learning rate
Для обучения используется оптимизатор Adam c параметрами learning rate , = 0.9, = 0.999, = . Используется размер батча 128: по одному элементу батча на каждое ядро TPU. Learning rate линейно растет («warm-up») в течение первых 128 тысяч батчей и умножается на 0.95 после 6.4 миллионов батчей. На втором этапе обучения learning rate уменьшается в 2 раза.
Для стабилизации обучения используется gradient clipping по глобальной L2-норме 0.1, независимо по каждому примеру в батче. Можно предположить, что за счет очень маленького значения L2-нормы gradient clipping оказывает принципиальное влияние на обучение.
Инициализация весов
Линейные слои с функцией активации ReLU инициализируются методом He normal.
Линейные слои, используемые для проекции векторов в keys, queries, values инициализируются методом Glorot uniform.
Другие линейные слои инициализируются методом LeCun normal.
В каждом residual-блоке последний слой инциализируется нулями.
Выходные слои AlphaFold также инициализируются нулями.
Линейные слои с сигмоидой, используемые в gating (см. раздел «Evoformer») инициализируются нулями, при этом их bias’ы инициализируются единицами. Тем самым обеспечивается «открытое состояние» всех гейтов в начале обучения.
Dropout
В evoformer-блоках (см. алгоритм 6) используются модификации dropout: row-wise и column-wise. Например, , действующий на массив MSA representation размером (, , ) (без учета размера батча), генерирует случайную бинарную маску размером (1, , ), и перемножает ее с массивом MSA representation.
Уменьшение потребления памяти
При обучении требуется сохранять выходные данные промежуточных слоев, чтобы затем выполнить обратный проход и рассчитать градиенты. Однако в сети AlphaFold есть места, где промежуточные данные имеют очень большой размер. Например, в triangular self-attention размер промежуточного массива (см. алгоритм 13) пропорционален третьей степени количества позиций (). Хранение этого массива в формате bfloat16 (2 байта) для всех 48 слоев evoformer-стека потребовало бы 20 гигабайт памяти для одного обучающего примера.
Для сокращения объема требуемой памяти авторы используют технику gradient checkpointing, иначе называемую rematerialization. При этом сохраняются только массивы, передаваемые между evoformer-блоками. Когда обратное распространение ошибки доходит до -го блока, заново делается прямой проход по этому блоку и рассчитываются градиенты. Таким образом, потребление памяти сокращается в десятки раз, а время одного шага обучение увеличивается лишь на 33%.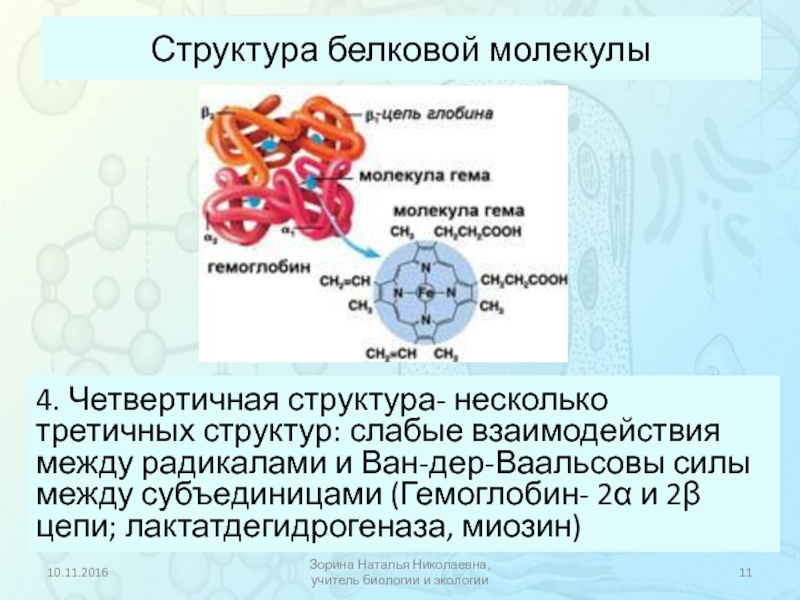
Еще одна техника сокращения потребления памяти применяется при инференсе. Если белок, для которого требуется рассчитать структуру, имеет очень большую длину (например, один из белков имеет длину 2180), то в каждом evoformer-блоке массив будет иметь размер 154 гигабайта. Для уменьшения объема требуемой памяти этот массив рассчитывается не целиком, а по частям, благодаря аддитивности операции triangular self-attention.
We identify a ‘batch-like’ dimension where the computation is independent along that dimension. We then execute the layer one ‘chunk’ at a time, meaning that only the intermediate activations for that chunk need to be stored in memory at a given time.
Авторы ссылаются на статью Reformer: The Efficient Transformer, где используется такой же подход.
Подробнее см. Supplementary Material, раздел 1.11.8
Self-distillation
Обучив одну модель на доступных исходных данных (MSA-последовательностях и 3D-структурах известных белков), авторы затем обучают следующую модель на датасете, состоящем на 75% из 3D-структур, предсказанных предыдущей моделью.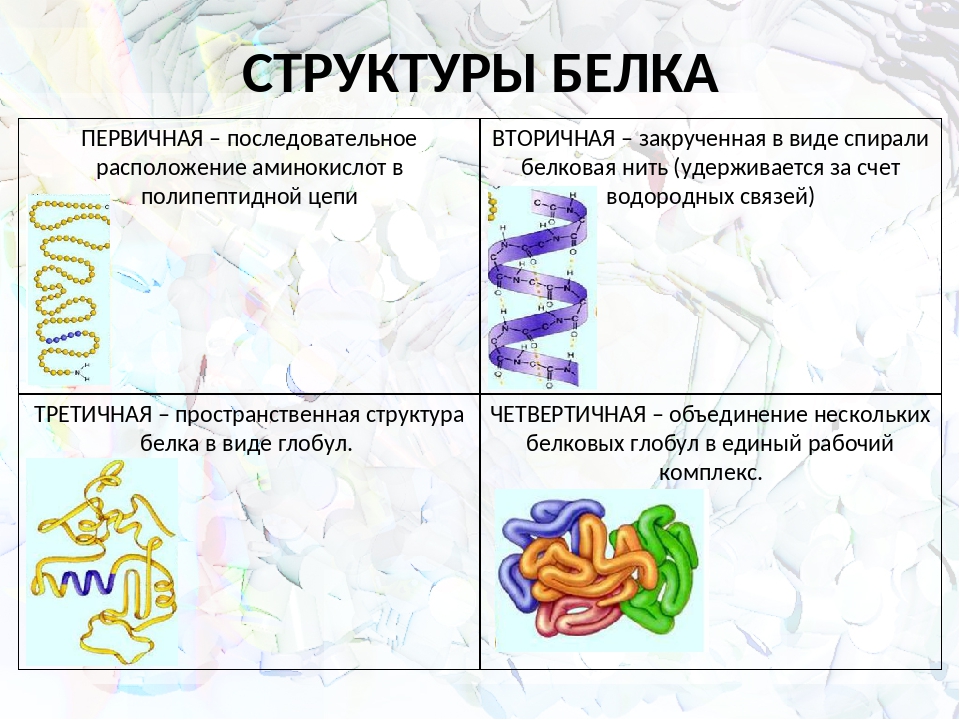 Такой способ обучения, называемый noisy-student self-distillation, ранее применялся в сверточных сетях (Self-training with noisy student improves imagenet classification, 2019), и также здесь прослеживается связь с более ранней работой Do Deep Nets Really Need to be Deep? (2013), где использовался аналогичный подход.
Такой способ обучения, называемый noisy-student self-distillation, ранее применялся в сверточных сетях (Self-training with noisy student improves imagenet classification, 2019), и также здесь прослеживается связь с более ранней работой Do Deep Nets Really Need to be Deep? (2013), где использовался аналогичный подход.
Подробнее см. Supplementary Material, раздел 1.3
Дополнительные разделы статьи
Ablation studies
Авторы пробуют удалять из AlphaFold различные компоненты и исследуют как это скажется на точности предсказаний. О результатах таких экспериментов уже упоминалось в разных частях этого обзора.
Network probing
В течение трех recycling iterations информация трижды проходит через каждый из 48 evoformer-блоков. Авторы присоединяют по одному дополнительному structure module к выходу каждого evoformer-блока на каждой итерации и обучают эти модули, блокируя протекание градиента из них в evoformer-блоки. Тем самым, дополнительные structure modules учатся предсказывать 3D-структуру по промежуточным выходным данным сети AlphaFold, при этом эти дополнительные модули не оказывают влияния на обучения основной части сети.
Таким образом, при инференсе мы получаем не только финальное предсказание 3D-структуры, но и 192 дополнительных предсказания – по одному для каждого evoformer-блока в каждой итерации. На рис. 26 показана точность предсказаний по метрике global distance test (GDT) для трех белков. Как видим, для простых белков AlphaFold почти сразу находит верную структуру, а для более сложных белков требуется несколько end-to-end запусков сети.
Рис. 26. Точность промежуточных предсказаний AlphaFold.Приведенные ниже видео показывают эволюцию предсказанных структур в ходе recycling iterations (номер кадра соответствует позиции по горизонтальной оси на рис. 26).
Резюме
В этом разделе еще раз суммирована основная информация из данного обзора.
Для предсказания структуры белка мы в первую очередь ищем в базе данных другие похожие белки. Из этих белков составляется таблица MSA (multiple sequence alignment). Белки в таблице MSA как правило являются эволюционными родственниками.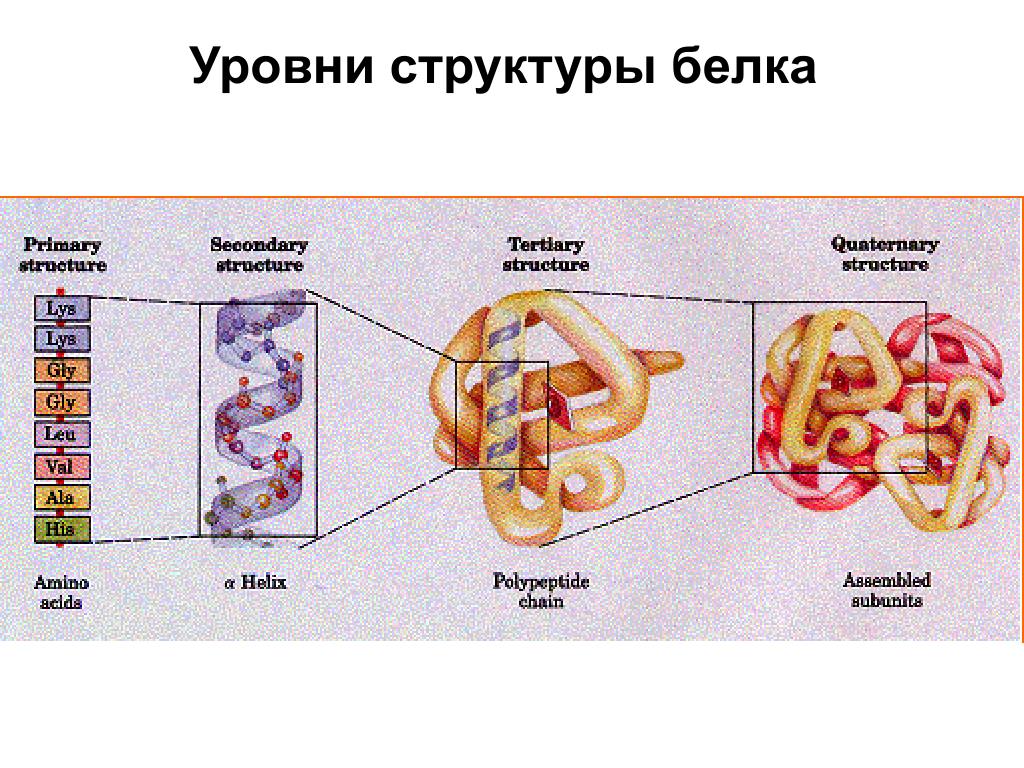 Мутации обычно затрагивают участки белка, не критично важные для сохранения его структуры, либо происходят парные мутации – в противном случае белок теряет свою функцию, и организм отсеивается естественным отбором. Благодаря этому по таблице MSA можно строить гипотезы о структуре белка. Если же в таблице MSA есть белки с уже известной 3D-структурой, то можно использовать и эту информацию («шаблоны»).
Мутации обычно затрагивают участки белка, не критично важные для сохранения его структуры, либо происходят парные мутации – в противном случае белок теряет свою функцию, и организм отсеивается естественным отбором. Благодаря этому по таблице MSA можно строить гипотезы о структуре белка. Если же в таблице MSA есть белки с уже известной 3D-структурой, то можно использовать и эту информацию («шаблоны»).
Входными данными для AlphaFold являются:
Аминокислотная последовательность исследуемого белка
MSA-таблица исследуемого белка
Набор шаблонов (опционально)
Архитектура AlphaFold состоит из трех последовательно соединенных блоков.
В первом блоке (feature embeddings) входные данные переводятся в эмбеддинги. Выходными данными блока являются:
MSA representation – абстрактное представление MSA-таблицы. Массив с тремя осями (без учета размера батча): номер последовательности, номер позиции в белке, номер элемента эмбеддинга.

Pair representation – абстрактное представление взаимодействий между каждой парой позиций в белке. Массив с тремя осями: номер первой позиции, номер второй позиции, номер элемента эмбеддинга.
Pair representation можно считать обобщением массива попарных расстояний (дистограммы), и интерпретировать как эмбеддинги ребер в полносвязном ориентированном графе между всеми позициями в белке.
Второй блок (evoformer stack) состоит из 48 последовательно соединенных evoformer-блоков. Каждый evoformer-блок состоит из последовательности операций, и вокруг каждой операции проброшен skip connection. Большая часть операций, выполняемых в evoformer-блоке – это multi-head self-attention, работающий по строкам или по столбцам массивов MSA representation и pair representation. Дополнительно выполняются следующие операции:
Третий блок, structure module, создает трехмерную структуру, используя в качестве входных данных:
Single representation – первую строку MSA representation, полученную на выходе из evoformer stack.
 Ее можно рассматривать как вектор-эмбеддинг для каждой позиции в белке.
Ее можно рассматривать как вектор-эмбеддинг для каждой позиции в белке.Pair representation, полученный на выходе из evoformer stack. Вектор-эмбеддинг для каждой пары позиций в белке.
Трехмерная структура белка представляется как массив backbone frames, называемый также «residue gas» — набор локальных ортонормированных систем координат, связанных с каждой позицией в белке. На residue gas не накладывается явного ограничения, что позиции должны быть объединены в цепочку – это правило сеть выучивает самостоятельно. Все координаты инициализируются нулями.
Блок structure module устроен достаточно сложно. В нем используется invariant point attention (IPA), в котором single representation обновляется под действием backbone frames, но инвариантно к смене глобальной системы координат в backbone frames. Функция потерь FAPE также инвариантна к смене глобальной системе координат (повороту, смещению) как в предсказанной, так и в эталонной структуре.
Помимо основной функции потерь минимизируется набор дополнительных функций потерь (auxiliary losses), например loss предсказания гистограммы по pair representation и loss предсказания пропусков в MSA representation (как в BERT). Для улучшения качества предсказания модель запускается несколько раз (recycling), каждый раз получая на вход предыдущее предсказание (вместе с шаблонами 3D-структуры).
Авторы отмечают, что радикальное упрощение structure module (включая удаление IPA) лишь немного ухудшает качество предсказания, тогда как минимизация только loss предсказания дистограммы существенно ухудшает качество. Из этого можно сделать вывод, что важна не столько сложность блока structure module, сколько его наличие и распространение градиента из этого модуля в evoformer stack.
Данный обзор первоначально размещен на моем сайте www.generalized.ru, там вы можете найти обзоры и на другие статьи.
Влияние посттрансляционной модификации митохондриальных и цитоплазматических белков плаценты на развитие задержки роста плода
Задержка роста плода (ЗРП) является тяжелой акушерской патологией, приводящей к высокой перинатальной заболеваемости и смертности. Несмотря на большое количество работ, посвященных ЗРП, механизмы ее развития недостаточно исследованы, что отражается на эффективности лечения. Поскольку нарушение роста плода во многом зависит от повреждения функций плаценты, изменение метаболических процессов в этом органе, особенно на молекулярном уровне, может играть ведущую роль в развитии ЗРП.
Несмотря на большое количество работ, посвященных ЗРП, механизмы ее развития недостаточно исследованы, что отражается на эффективности лечения. Поскольку нарушение роста плода во многом зависит от повреждения функций плаценты, изменение метаболических процессов в этом органе, особенно на молекулярном уровне, может играть ведущую роль в развитии ЗРП.
ЦЕЛЬ ИССЛЕДОВАНИЯ
Изучить посттрансляционные модификации митохондриальных и цитоплазматических белков плаценты при ЗРП и оценить их роль в формировании этого осложнения беременности.
МАТЕРИАЛ И МЕТОДЫ
В обсервационное одноцентровое одномоментное контролируемое исследование включены 27 клинически здоровых женщин с неосложненной беременностью (контрольная группа) и 30 женщин с симметричной формой ЗРП (основная группа). Использованы клинические, клинико-лабораторные и функциональные методы обследования. Материалом для исследования служила ткань плаценты. Для выделения белков субклеточных фракций плаценты использовали метод дифференциального ультрацентрифугирования. Показатели посттрансляционной модификации белков (амидирования, аминирования, карбонилирования, фосфорилирования) оценивали с помощью спектрофотометрических и радиоизотопных методов.
Показатели посттрансляционной модификации белков (амидирования, аминирования, карбонилирования, фосфорилирования) оценивали с помощью спектрофотометрических и радиоизотопных методов.
РЕЗУЛЬТАТЫ
Женщины обеих групп статистически значимо не различались по акушерскому и соматическому анамнезу. При ЗРП, развившейся на фоне дисфункции плаценты, имеются значительные изменения плацентарных белков: снижается интенсивность их циклонуклеотид-зависимого фосфорилирования, амидирования, аминирования и повышается спонтанное и металл-катализируемое окислительное карбонилирование. Степень этих изменений зависит от циклического нуклеотида, субклеточной локализации белков, особенностей изученных функциональных групп. Установленные нарушения отражаются на разных уровнях структуры белков и, следовательно, на их регуляторных свойствах.
ВЫВОДЫ
Выявленные нарушения структуры митохондриальных и цитоплазматических белков плаценты, выполняющих многочисленные регуляторные функции, могут быть важными звеньями в цепи молекулярных нарушений при задержке роста плода.
Заполните таблицу. Особенности строения белковой молекулы Структура молекулы белка Особенности строения
Особенности строения белковой молекулы
Структура белкововой молекулы — сложная пространственная структура, обладающая первичным, вторичным, третичным и четвертичным уровнями организации. Особенности структурной организации белковой молекулы определяются первичным уровнем ее организации.
Первичная структура белковой молекулы — полипептидная цепь с линейной последовательностью амнокислот, связанных между собой за счет пептидной связи. Первичная структура белка наиболее прочная из всех. В отношений всех свойств, которыми будет обладать белковая молекула, эта структура является определящюей. Все остальные структурные уровни организации образуются в соответствии с особенностями строения первичного уровня по принципу самосборки. Внешние факторы не оказывают влияние на этот процесс.
Вторичная структура белковой молекулы — структура белковой молекулы, образующаяся за счёт скручивания линейной последовательности аминокислот первичной структуры с образованием спирали, многочисленные витки которой связаны между собой водородными связями.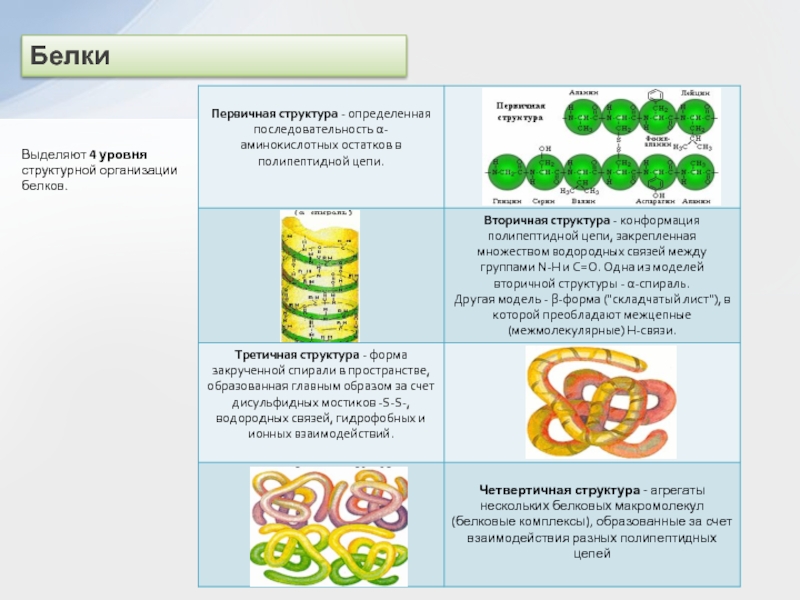
Третичная структура белковой молекулы — структура белковой молекулы, образующаяся за счет наложения одних частей спирали белковой молекулы на другие, формирование между этими частями различного рода связей: водородных ковалентных ионных, дисульфидных (при наличии аминокислоты цистин), гидрофобных. Третичная структура имеет вид глобулы. При третичном уровне организации белковой молекулы возможность принимать участие в химических реакциях, проявлять химическую активность остается только у тех аминокислотных остатков, которые имеют поверхностное расположение.
Четвертичная структура белковой молекулы — структура белковой молекулы, представляющая собой сложную пространственную организацию нескольких полипептидных цепей, связанных между собой за счет различных химических связей. Эти связи аналогичны таковым в третичном уровне организации белковой молекулы. Полипептидные цепи, принимающие участие в образовании четвертичной структы белковой молекулы, могут быть одинаковыми или иметь различное строение.
Структура белка — Химия LibreTexts
- Последнее обновление
- Сохранить как PDF
Вторичная структура относится к форме сворачивающегося белка, обусловленной исключительно водородными связями между амидной и карбонильной группами его основной цепи.Вторичная структура не включает связи между R-группами аминокислот, гидрофобные взаимодействия или другие взаимодействия, связанные с третичной структурой. Двумя наиболее часто встречающимися вторичными структурами полипептидной цепи являются α-спирали и бета-складчатые листы. Эти структуры являются первыми основными шагами в сворачивании полипептидной цепи, и они устанавливают важные топологические мотивы, которые определяют последующую третичную структуру и окончательную функцию белка.
- Сворачивание белков
- Вторичная структура: α-спирали
- α-спираль представляет собой правозакрученный виток аминокислотных остатков в полипептидной цепи, обычно содержащий от 40 до 40 остатков.Этот виток удерживается вместе водородными связями между кислородом C=O в верхнем витке и водородом NH в нижнем витке.
- Вторичная структура: β-складчатый лист
- Эта структура возникает, когда два (или более, например, ψ-петля) сегмента полипептидной цепи накладываются друг на друга и образуют ряд водородных связей друг с другом. Это может происходить при параллельном расположении или при встречно-параллельном расположении. Параллельное и антипараллельное расположение является прямым следствием направленности полипептидной цепи.
- Вторичная структура: α-складчатый лист
- Структура, аналогичная бета-складчатому листу, представляет собой α-складчатый лист.
 Эта структура энергетически менее выгодна, чем бета-складчатый лист, и довольно редко встречается в белках. Альфа-складчатый лист характеризуется выравниванием карбонильных и аминогрупп; все карбонильные группы выровнены в одном направлении, а все группы NH выровнены в противоположном направлении.
Эта структура энергетически менее выгодна, чем бета-складчатый лист, и довольно редко встречается в белках. Альфа-складчатый лист характеризуется выравниванием карбонильных и аминогрупп; все карбонильные группы выровнены в одном направлении, а все группы NH выровнены в противоположном направлении.
- Структура белков
- На этой странице объясняется, как аминокислоты объединяются в белки, и что подразумевается под первичной, вторичной и третичной структурой белков.Четвертичная структура не раскрыта. Это относится только к белкам, состоящим более чем из одной полипептидной цепи.
Миниатюра: Структура гемоглобина человека. Субъединицы белков α и β отмечены красным и синим цветом, а железосодержащие гемовые группы – зеленым. (CC BY-SA 3.0; Zephyris).
Вторичная структура белка — обзор
Предсказание вторичной структуры было выполнено для определения структурной значимости целевых последовательностей с использованием PSIPRED, основанного на словаре вторичной структуры белка (Kabsch and Sander, 1983).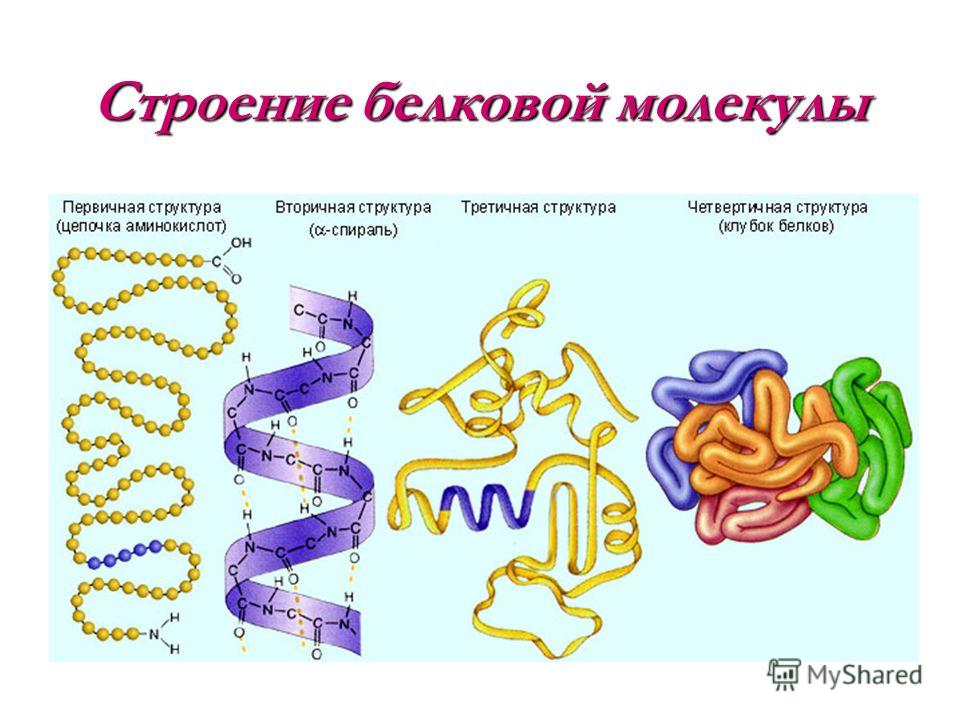 Компания MODELLER сконструировала трехмерные модели пептида RIPL для выбора наилучшей модели с наивысшим показателем достоверности. Доступ к этим инструментам осуществлялся через локальный сервер метапотоков (http://zhanglab.ccmb.med.umich.edu/LOMETS/) (Wu and Zhang, 2007). Структура пептида RIPL и комплекса Hpn была предсказана и описана с помощью ZDOCK, алгоритма стыковки белков (http://zlab.umassmed.edu/zdock/) (Pierce et al., 2011).
Компания MODELLER сконструировала трехмерные модели пептида RIPL для выбора наилучшей модели с наивысшим показателем достоверности. Доступ к этим инструментам осуществлялся через локальный сервер метапотоков (http://zhanglab.ccmb.med.umich.edu/LOMETS/) (Wu and Zhang, 2007). Структура пептида RIPL и комплекса Hpn была предсказана и описана с помощью ZDOCK, алгоритма стыковки белков (http://zlab.umassmed.edu/zdock/) (Pierce et al., 2011).
Вторичная и третичная структуры пептида RIPL были проверены с использованием соответствующих программ.Новый CPHP несет два основных домена IPL для направленного действия и R8 для проникающего действия в клетку. Аналоги IPL были выбраны случайным образом с помощью фагового дисплея в клетках PC3, трансфицированных Hpn, на основании их общей гомологии во вторичной структуре белковых последовательностей (Kelly et al., 2008). Чтобы исследовать структурное сходство между пептидом RIPL и аналогами IPL, мы предсказали вторичную структуру пептида RIPL, используя метод PSIPRED, в котором вторичная структура обозначена как C (виток), E (цепь) и H (спираль). Последовательность аналогов IPL содержит общую структуру связанной катушки и нити (CCEE) (таблица 5.3). В пептиде RIPL С-конец пептида IPL был соединен с пептидом R8. Несмотря на связь pArg, эта общая вторичная структура была сохранена, чтобы гарантировать, что свойства нацеливания Hpn не будут затруднены. Мы обнаружили, что RIPL-пептид с флуоресцентной меткой (RIPL-FITC) демонстрировал большее клеточное поглощение клетками Hpn(+), чем поглощение при использовании только FITC-декстрана, что указывает на то, что аффинность Hpn сохранялась независимо от связи R8.Предлагаемые третичные структуры пептида RIPL и Hpn показаны на рис. 5.2.
Последовательность аналогов IPL содержит общую структуру связанной катушки и нити (CCEE) (таблица 5.3). В пептиде RIPL С-конец пептида IPL был соединен с пептидом R8. Несмотря на связь pArg, эта общая вторичная структура была сохранена, чтобы гарантировать, что свойства нацеливания Hpn не будут затруднены. Мы обнаружили, что RIPL-пептид с флуоресцентной меткой (RIPL-FITC) демонстрировал большее клеточное поглощение клетками Hpn(+), чем поглощение при использовании только FITC-декстрана, что указывает на то, что аффинность Hpn сохранялась независимо от связи R8.Предлагаемые третичные структуры пептида RIPL и Hpn показаны на рис. 5.2.
Таблица 5.3. Вторичная структура пептидов Прогнозируемая по методу PSIPRED
| пептидной последовательности | Вторичная структура |
|---|---|
| IPL аналоги | |
| IPLVVPL | CCEECCC |
| IPLWVPL | CCEECCC |
| IPLVLVPL | |
| IPLVVPLGGSCK CCEEEECCCCCC | |
| RIPL пептид | |
| IPLVVPLRRRRRRRRC CCEEEHHHHHHHHCCC | |
Перепечатывается из Kang, M. H., Park, MJ, Yoo, HJ, Kwon, YH, Lee, SG, Kim, SR, Yeom, DW, Kang, MJ, Choi, YW, 2014. Липосомы, конъюгированные с пептидом RIPL (IPLVVPLRRRRRRRRC) для улучшенной внутриклеточной доставки лекарств к экспрессирующим хепсин раковым клеткам. Евро. Дж. Фарм. Биофарм. 87, 489–499, с разрешения Elsevier.
H., Park, MJ, Yoo, HJ, Kwon, YH, Lee, SG, Kim, SR, Yeom, DW, Kang, MJ, Choi, YW, 2014. Липосомы, конъюгированные с пептидом RIPL (IPLVVPLRRRRRRRRC) для улучшенной внутриклеточной доставки лекарств к экспрессирующим хепсин раковым клеткам. Евро. Дж. Фарм. Биофарм. 87, 489–499, с разрешения Elsevier.
Рисунок 5.2. Предлагаемые третичные структуры пептида RIPL и Hpn
W., 2014. Липосомы, конъюгированные с пептидом RIPL (IPLVVPLRRRRRRRRC), для усиленной внутриклеточной доставки лекарств к раковым клеткам, экспрессирующим хепсин. Евро. Дж. Фарм. Биофарм. 87, 489–499, с разрешения Elsevier.Белковая структура | Биониндзя
Понимание:
• Последовательность и количество аминокислот в полипептиде является первичной структурой
Первичная (1º) структура
- Первый уровень структурной организации в белке – это порядок/последовательность аминокислот , которые составляют полипептидную цепь
- Первичная структура образована ковалентными пептидными связями между амино- и карбоксильные группы соседних аминокислот
- Первичная структура контролирует все последующие уровни организации белка, поскольку определяет характер взаимодействий между R-группами различных аминокислот
Понимание:
• Вторичная структура представляет собой образование α-спиралей и β-складок, стабилизированных водородными связями
Структура вторичного (2º)
- Вторичная структура — это способ полипептидных складок в повторяющихся агрегат для формы α- Heatics и β-плиссированные листы
- Эта складка является результатом из водородная связь между аминогруппами и карбоксильной группой несмежных аминокислот
- Последовательности, которые не образуют ни альфа-спирали, ни бета-складчатого листа, будут существовать в виде случайного клубка
- Вторичная структура обеспечивает полипептидную цепь уровень механической стабильности (благодаря наличию водородных связей)
- На рисунках альфа-спирали представлены в виде спиралей (фиолетовый; слева) и бета-складчатые листы в виде стрелок (синий; справа)
Понимание:
• Третичная структура представляет собой дальнейшую укладку полипептида, стабилизированную взаимодействиями между R-группами
Третичная (3º) структура
- Третичная структура представляет собой способ, которым полипептидная цепь закручивается и поворачивается, образуя сложную молекулярную форму (т.
 е. 3D-форма )
е. 3D-форма ) - Это вызвано взаимодействиями между группами R ; включая Н-связи, дисульфидные мостики, ионные связи и гидрофобные взаимодействия
- Важны относительные положения аминокислот (например, неполярные аминокислоты обычно избегают воздействия водных растворов)
- Третичная структура может быть важна для функции белка (например, специфичность активного центра в ферментах)
Понимание:
• Четвертичная структура существует в белках с более чем одной полипептидной цепью
Четвертичная (4º) структура
- Несколько полипептидов или простетических групп могут взаимодействовать с образованием одного, более крупного, биологически активного белка (четвертичная структура)
- Простетическая группа представляет собой неорганическое соединение или функция (т.г. гемовая группа в гемоглобине)
- Белок, содержащий простетическую группу, называется конъюгированным белком
- Четвертичные структуры могут удерживаться вместе различными связями (подобно третичной структуре)
Краткий обзор четырех уровней белковой структуры
уровней организации белка
уровень организации белка Уровни организации белков
A 2014 Foundations of Medicine eLAB
Первичная структура белка определяется как аминокислотная последовательность его полипептидной цепи; вторичная структура — локальное пространственное расположение атомов остова (основной цепи) полипептида; третичная структура относится к трехмерной структуре всей полипептидной цепи; а четвертичная структура представляет собой трехмерное расположение субъединиц в мультисубъединичном белке. В этой серии страниц мы исследуем различные уровни организации белка. Мы также по-разному рассматриваем структуры — скелет Cα, шарико-палочка, CPK, лента, заполнение пространства — а также цвет используется для выделения различных аспектов аминокислот, структуры и т. д. По мере прохождения этого модуля обратите внимание на эти аспекты.
В этой серии страниц мы исследуем различные уровни организации белка. Мы также по-разному рассматриваем структуры — скелет Cα, шарико-палочка, CPK, лента, заполнение пространства — а также цвет используется для выделения различных аспектов аминокислот, структуры и т. д. По мере прохождения этого модуля обратите внимание на эти аспекты.
Этот модуль включает ссылки на KiNG (Kinemage, Next Generation), который отображает трехмерные структуры в анимированном интерактивном формате.Эти «кинемажи» (кинетические изображения) можно вращать, перемещать и масштабировать, а части можно скрывать или отображать. Первоначально Kinemages были реализованы под эгидой Фонда инновационных технологий и Белкового общества, а программированием и обслуживанием занимались Дэвид С. Ричардсон и Джейн С. Ричардсон.
Ссылка : «КИНЕМАЖ: ИНСТРУМЕНТ НАУЧНОЙ КОММУНИКАЦИИ» Д.К. Ричардсон и Дж.С. Richardson (1992) Protein Science 1: 3-9.Также Тенденции в биохимии. науч. (1994) 19: 135-8.
науч. (1994) 19: 135-8.
Текст адаптирован из : Demo5_4a.kin
Первичная структура пептида или белка представляет собой линейную последовательность его аминокислот (АА). По соглашению первичная структура белка читается и записывается от амино-конца (N) к карбокси-концу (С). Каждая аминокислота соединена со следующей пептидной связью.
В то время как первичная структура описывает последовательность аминокислот, образующих пептидную цепь, вторичная структура относится к локальному расположению цепи в пространстве.В белках идентифицировано несколько общих вторичных структур. Они будут описаны в следующих разделах и визуализированы с использованием программного обеспечения KiNG, упомянутого ранее.
Чтобы загрузить Java-апплет KiNG, просто нажмите здесь. После загрузки этой страницы Java-апплет KiNG должен появиться автоматически. Если вам нужна информация об использовании King, пожалуйста, наведите курсор сюда.
Альфа-Хеликс
Альфа-спираль – это элемент вторичной структуры, в котором цепь аминокислот расположена по спирали.На приведенном выше кинемаже показана отдельная альфа-спираль, если смотреть с N-концевого конца, она напоминает «винтовое колесо» (см. Рисунок ниже). Атомы O и N основной цепи спирали показаны красными и синими шариками соответственно. Нецелое повторение 3,6 остатка на виток альфа-спирали означает, что Cα последовательных витков смещены примерно наполовину, что придает основной цепи характерный вид 7-конечной звезды на виде сбоку. Обратите внимание, что связи Cα-Cβ не направлены радиально от оси спирали, а «вертятся» вдоль линии одного из соседних пептидов, придавая боковым цепям асимметричное начало.
Гидрофобные боковые цепи показаны зеленым цветом, полярные – небесно-голубым, а заряженные – красным. Их можно включить, установив флажок «side ch». Теперь ВКЛЮЧИТЕ и ВЫКЛЮЧИТЕ различные группы и наборы дисплеев, нажав соответствующую кнопку.
Когда вы включали различные типы сайдчейнов, что вы наблюдали? Вы заметили, что у спирали одна сторона преимущественно с полярными остатками, а другая с преимущественно гидрофобными остатками? Это типичная глобулярно-белковая спираль; в его нативной конфигурации полярные остатки будут обращены к растворителю, а гидрофобные остатки будут обращены внутрь белка.В меню просмотра в KiNG выберите View2 или View3, чтобы увидеть больше структуры.
На рисунке слева показано изображение аминокислотной последовательности в виде спирального колеса, если смотреть вниз по оси альфа-спирали, перпендикулярной странице. Аминокислотные остатки пронумерованы от ближайших к наиболее удаленным и расположены в виде идеальной альфа-спирали с 3,6 остатками на полный оборот. Этот рисунок представляет собой снимок Java-апплета, написанного Эдвардом К. О’Нилом и Чарльзом М. Гришамом (Университет Вирджинии в Шарлоттсвилле, Вирджиния).
В KiNG выберите View4 для крупного плана сбоку, где спиральные водородные связи (Н-связи) выделены коричневым цветом. Включите «Hbonds» на панели кнопок, чтобы увидеть H-связи в коричневом цвете. Нажмите на атомы основной цепи на любом конце одной из Н-связей, чтобы убедиться, что альфа-спиральная модель Н-связи действительно идет от донора NH в остатке i к акцептору O в остатке i-4 (как показано на рисунке). рисунок справа). Проверьте, имеет ли эта альфа-спираль 3,6 остатка на виток. Если бы вы были уверены, подъем на полный оборот равен 5.4 ангстрема (В).
Включите «Hbonds» на панели кнопок, чтобы увидеть H-связи в коричневом цвете. Нажмите на атомы основной цепи на любом конце одной из Н-связей, чтобы убедиться, что альфа-спиральная модель Н-связи действительно идет от донора NH в остатке i к акцептору O в остатке i-4 (как показано на рисунке). рисунок справа). Проверьте, имеет ли эта альфа-спираль 3,6 остатка на виток. Если бы вы были уверены, подъем на полный оборот равен 5.4 ангстрема (В).
правосторонние. Чтобы увидеть, что это правша, держите правую руку так, чтобы большой палец был направлен вверх, а пальцы были свободно согнуты; стараясь соответствовать спирали спирали, медленно двигайтесь в направлении, указанном большим пальцем, и скручивайтесь вдоль линии пальцев, как бы затягивая винт. Когда это движение соответствует спирали позвоночника, если оно выполняется правой рукой, тогда спираль правосторонняя.
Чтобы измерить углы φ,psi для примера спирали KiNG, включите «Измерить угол и двугранный угол» в раскрывающемся меню «Инструменты». Начните с щелчка по карбонильному атому C вверху, затем по следующему N, затем по Cα и снова по C; в этой точке информационная линия покажет двугранный угол, который является углом фи центральной связи N-Cα этих 4 атомов. Для правосторонней альфа-спирали она должна находиться в диапазоне от -50 до -80 градусов. Нажмите на следующий N, и вы получите угол psi, который должен быть между -25 и -60 градусами. Продолжайте движение вниз по спирали, получая омегу (около 180 градусов), фи, пси и т. д. Эти спиральные значения фи, пси находятся в густонаселенной области в левом нижнем углу графика Рамачандрана (показано справа).
Начните с щелчка по карбонильному атому C вверху, затем по следующему N, затем по Cα и снова по C; в этой точке информационная линия покажет двугранный угол, который является углом фи центральной связи N-Cα этих 4 атомов. Для правосторонней альфа-спирали она должна находиться в диапазоне от -50 до -80 градусов. Нажмите на следующий N, и вы получите угол psi, который должен быть между -25 и -60 градусами. Продолжайте движение вниз по спирали, получая омегу (около 180 градусов), фи, пси и т. д. Эти спиральные значения фи, пси находятся в густонаселенной области в левом нижнем углу графика Рамачандрана (показано справа).
Таким образом, идеальная альфа-спираль обладает следующими свойствами:
- Делает один оборот каждые 3,6 остатка;
- Поднимается примерно на 5,4 Â с каждым оборотом;
- Это правосторонняя спираль;
- Он удерживается вместе водородными связями между C=O остатка i и NH остатка i+4;
- Обычно слегка изогнут.
Некоторые общие свойства альфа-спиралей:
- Средняя длина альфа-спирали составляет 10 остатков (15 Â), хотя в стандартном глобулярном белке длина альфа-спирали может варьироваться от 4 до 40 остатков.

- Все остатки, участвующие в альфа-спирали, имеют одинаковые углы (phi,psi). Эти углы, равные примерно -60 и -50, взяты из нижнего левого квадранта графика Рамачандрана.
- Некоторые аминокислоты предпочтительно расположены в альфа-спирали. Остатки, такие как Ala, Glu, Leu и Met, имеют высокую склонность к участию в спирали, в то время как остатки, такие как Pro и Gly, имеют небольшую такую тенденцию. Особый интерес представляет пролин, который не может вписаться в спираль и создает изгиб.
- Спираль имеет общий дипольный момент, который представляет собой векторную сумму выровненных дипольных моментов отдельных пептидных связей. Положительный полюс находится на N-конце, а отрицательный полюс — на С-конце. Иногда этот диполь играет функциональную роль.
Некоторый текст адаптирован из : Приложение Kinemage к Branden & Tooze «Введение в структуру белка», глава 2 — МОТИВЫ СТРУКТУРЫ БЕЛКА Джейн С. и Дэвида С. Ричардсона.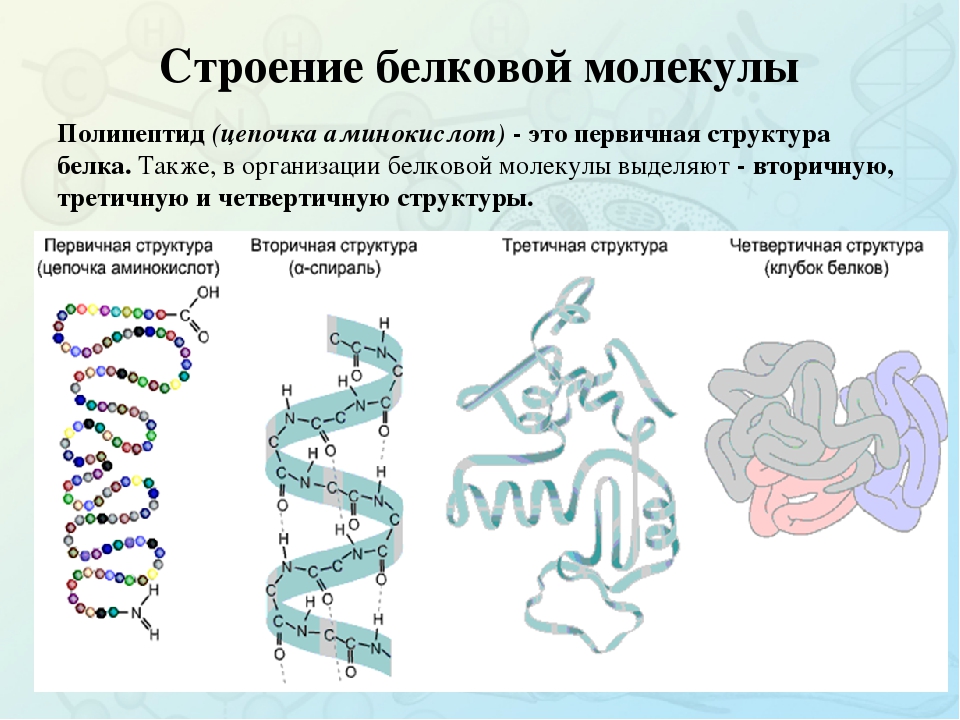
Бета-цепь представляет собой элемент вторичной структуры, в котором белковая цепь является почти линейной.Соседние бета-нити могут образовывать водородные связи с образованием бета-листа (также называемого бета-складчатым листом). Участвующие бета-цепи не являются непрерывными в первичной последовательности и даже не должны быть близки друг к другу в последовательности, т. е. нити, образующие бета-лист, могут быть разделены в первичной структуре длинными последовательностями аминокислот, не входящих в состав листа. Примерно четверть всех остатков в типичном белке находится в бета-цепях, хотя это сильно различается между белками
. Чтобы просмотреть бета-лист в Java-апплете KiNG, нажмите здесь.Kinemage 1 показывает 6-цепочечный параллельный бета-лист из домена 1 лактатдегидрогеназы (файл 1LDM). Этот параллельный бета-лист с двойной обмоткой является наиболее распространенным паттерном складывания, встречающимся в известных белковых структурах. Эта «складка» также известна как «домен, связывающий нуклеотиды», потому что большинство примеров связывают мононуклеотид (например, FMN) или динуклеотид (например, NAD) около середины одного конца бета-листа. Лактатдегидрогеназа является классическим, первым обнаруженным примером структуры этого типа и имеет наиболее часто наблюдаемую топологию бета-соединений.
Лактатдегидрогеназа является классическим, первым обнаруженным примером структуры этого типа и имеет наиболее часто наблюдаемую топологию бета-соединений.
Обратите внимание, что водородные связи в этом параллельном слое наклонены в разных направлениях, а не перпендикулярно нитям, как мы увидим в антипараллельных слоях. Перетащите вправо или влево, чтобы лучше увидеть, что лист в целом скручивается. Этот поворот обычно описывается поворотом в ориентации плоскостей пептида (или плоскости Н-связи) по мере продвижения по цепи; по этому определению скручивание бета-листа всегда правостороннее, хотя и в разной степени. Щелкайте по атомам вдоль нити, чтобы узнать ее направление по остаточным числам, и убедитесь, что все шесть нитей действительно параллельны.Метки нитей показывают порядок последовательности нитей. Обратите внимание, что большинство последовательных пар расположены рядом друг с другом, и что цепочка начинается в середине, движется к одному краю, возвращается к середине и затем перемещается к другому краю. Есть три возможных способа сформировать бета-лист из бета-цепи, обсуждаемые ниже.
Есть три возможных способа сформировать бета-лист из бета-цепи, обсуждаемые ниже.
Типы бета-листов, наблюдаемые в белках
1) Параллельный бета-лист. Все скрепленные нити имеют одинаковое направление от N к C. В результате они должны быть разделены длинными участками последовательности.Водородные связи равноудаленные.
На рисунке слева показан трехцепочечный параллельный бета-лист из белка тиоредоксина. Три параллельные нити показаны как в мультяшном формате (слева), так и в виде палочек, содержащих атомы основной цепи N, CA, C и O’ (справа). Водородные связи обозначены стрелками, соединяющими донорный азот и акцепторный кислород. Нити нумеруются в соответствии с их относительным положением в полипептидной последовательности.
2) Антипараллельный бета-лист. Бета-цепи проходят в чередующихся направлениях и, следовательно, могут быть довольно близки на первичной последовательности.Расстояние между последовательными водородными связями то короче, то длиннее.
На рисунке справа показан трехцепочечный антипараллельный бета-лист из тиоредоксина. Три антипараллельные нити показаны как в мультяшном формате (слева), так и в виде палочек, содержащих атомы основной цепи N, CA, C и O’ (справа). Водородные связи обозначены стрелками, соединяющими донорный азот и акцепторный кислород. Нити нумеруются в соответствии с их относительным положением в полипептидной последовательности.
3) Смешанный бета-лист – смесь параллельных и антипараллельных водородных связей. Около 20% всех бета-листов являются смешанными.
Образцы водородных связей в смешанном бета-листе (рисунок слева). Здесь схематично изображен четырехнитевой бета-лист, содержащий три антипараллельные нити и одну параллельную. Водородные связи между антипараллельными нитями показаны красными линиями, между параллельными нитями — зелеными.
Некоторые из основных функций бета-листов включают:
- Расширенная конформация бета-цепи составляет около 3.
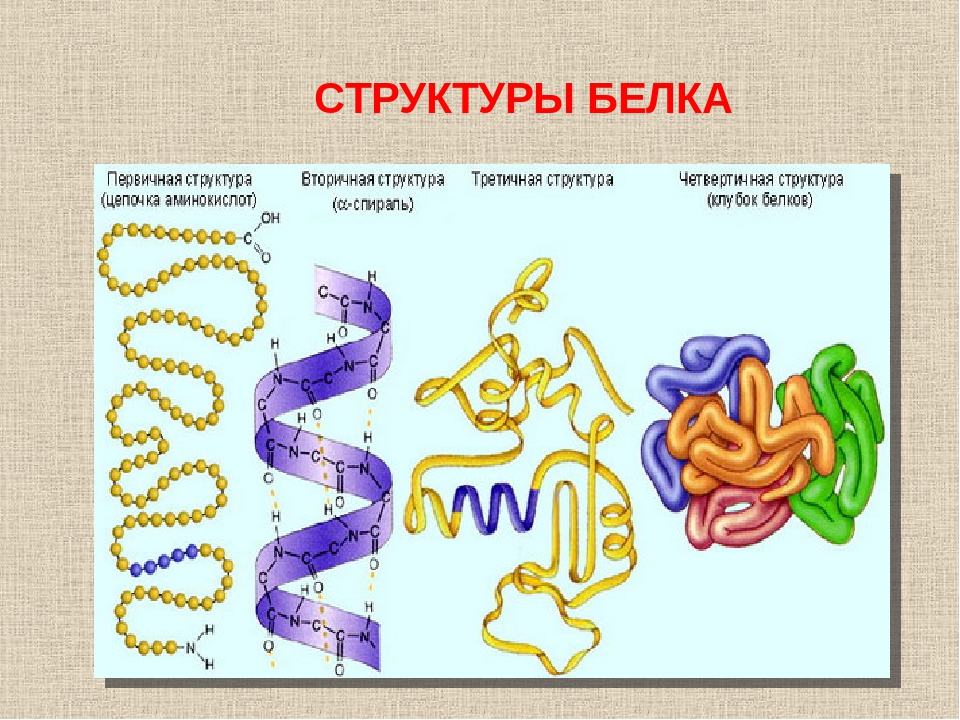 5 Â на остаток, а длина бета-цепи может достигать 35 Â.
5 Â на остаток, а длина бета-цепи может достигать 35 Â. - Общая геометрия листа не плоская, а складчатая, с чередованием атомов углерода Cα выше и ниже средней плоскости листа.
- Из-за хиральности аминокислот (L-аминокислоты) все бета-цепи имеют правый поворот, тогда как бета-лист имеет в целом левый поворот.
- Поскольку нити не обязательно должны быть соседними в последовательности, существует много возможных способов расположения нитей на листе, эти схемы называются топологиями и могут быть довольно сложными.
Включите боковые цепи в KiNG, чтобы проверить их расположение. Вдоль данной нити боковые цепи чередуются между одной стороной листа (золото) и другой (море или небо). На соседних нитях чередование совпадающее, так что боковые цепи образуют ряды, находящиеся в достаточно тесном контакте. На параллельном бета-листе геометрия такова, что боковые цепи с разветвленными бета-углеродами (Val, Ile или Thr) довольно благоприятно контактируют вдоль ряда; поскольку эти положения обычно скрыты и гидрофобны, в результате Val и Ile являются доминирующими остатками, обнаруживаемыми в этих положениях.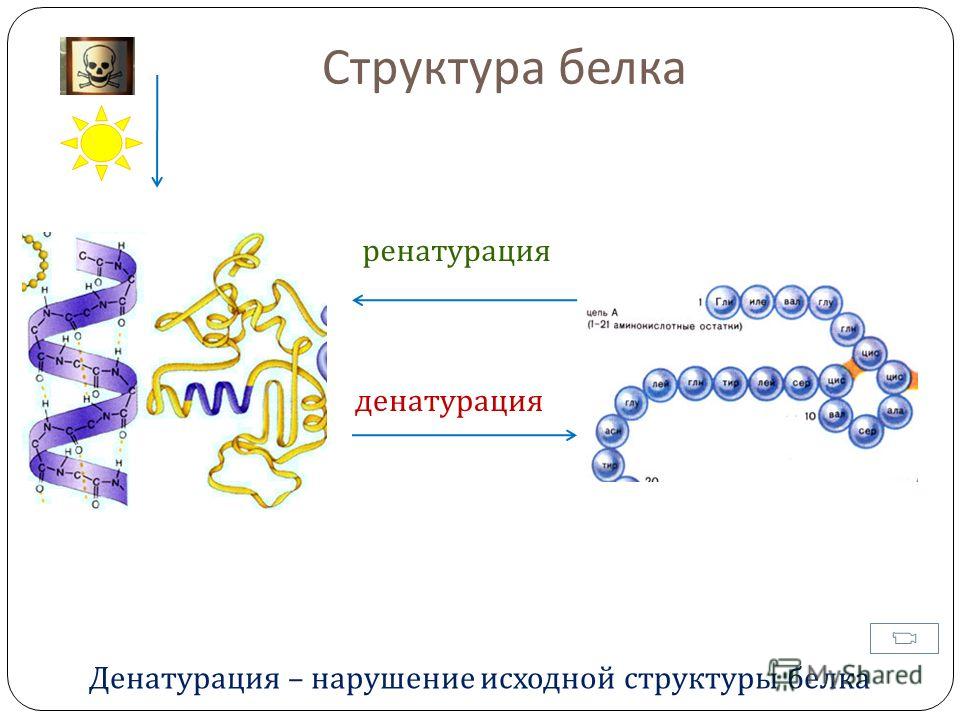 Краевые нити или самые концы данной нити могут подвергаться воздействию растворителя и часто имеют значительно более гидрофильные остатки (как, например, в ряду 0 здесь или Ser на нити 3).
Краевые нити или самые концы данной нити могут подвергаться воздействию растворителя и часто имеют значительно более гидрофильные остатки (как, например, в ряду 0 здесь или Ser на нити 3).
Некоторый текст адаптирован из : «Протеиновый турист: ПАРАЛЛЕЛЬНЫЕ АЛЬФА/БЕТА-БЕЛКИ С ДВУМЯ ОБОРОТКАМИ, ИЛИ НУКЛЕОТИД-СВЯЗЫВАЮЩИЕ ДОМЕНЫ» Дж.С. Ричардсон и Д. К. Ричардсон.
Бета-поворот
Повороты обычно возникают, когда белковой цепи необходимо изменить направление, чтобы соединить два других элемента вторичной структуры.Наиболее распространенным является бета-поворот, при котором изменение направления выполняется в пространстве четырех остатков. Некоторыми обычно наблюдаемыми особенностями бета-поворотов являются водородная связь между C=O остатка i и NH остатка i+3 (т. е. между первым и четвертым остатком поворота) и сильная тенденция к вовлечению глицина и / или пролин. Иногда вы будете слышать фразу «бета-шпилька», которую можно использовать для описания бета-поворота, соединяющего вместе две антипараллельные бета-нити. Бета-повороты подразделяются на многочисленные типы на основе деталей их геометрии.
Бета-повороты подразделяются на многочисленные типы на основе деталей их геометрии.
Гамма-повороты представляют собой повороты с тремя остатками, которые часто включают водородную связь между C=O остатка i и NH остатка i+2.
Случайная катушка
Некоторые участки белковой цепи не образуют регулярной вторичной структуры и не характеризуются какой-либо регулярной структурой водородных связей. Эти области известны как случайные клубки и находятся в белках в двух местах:
.- Концевые плечи — как на N-конце, так и на С-конце белка;
- Петли. Петли представляют собой неструктурированные области, находящиеся между обычными элементами вторичной структуры.
Случайные катушки могут иметь длину от 4 до 20 остатков, хотя длина большинства петель не превышает 12 остатков. Большинство петель подвергаются воздействию растворителя и имеют полярные или заряженные боковые цепи. В некоторых случаях петли играют функциональную роль, но во многих случаях их нет. В результате области петель часто плохо сохраняются (т.е. более склонны к изменениям) в процессе эволюции.
В результате области петель часто плохо сохраняются (т.е. более склонны к изменениям) в процессе эволюции.
Некоторый текст адаптирован из : «УПРАЖНЕНИЕ 3. ВТОРИЧНЫЕ СТРУКТУРЫ БЕЛКА» Ким М. Гернерт и Ким М.Кицлер.
Как мы узнали, порядок аминокислот является первичной структурой, и все остатки в полипептидной цепи имеют одинаковые атомы основной цепи. Что различается, так это боковые цепи (группы R). Определяют ли присутствующие специфические AA вторичную структуру? Как показано на рисунке, все аминокислоты можно найти во всех элементах вторичной структуры, но некоторые более или менее распространены в определенных элементах. Pro и Gly, например, не очень хороши для спиралей, но лучше подходят для бета-поворотов.Если мы сделаем еще один шаг и спросим, определяют ли комбинации 2, 3 или 4 аминокислот вторичную структуру, мы обнаружим более сильную корреляцию, но все же недостаточно сильную, чтобы надежно предсказать третичную структуру.
Белки имеются в изобилии во всех организмах и являются основой жизни. Разнообразие структуры белков лежит в основе очень большого спектра их функций: ферменты (биологические катализаторы), запасающие, транспортные, мессенджеры, антитела, регуляторные и структурные белки.
Белки представляют собой линейные гетерополимеры фиксированной длины; то есть один тип белка всегда имеет одинаковое количество и состав АК, но разные белки могут иметь от 100 до более 1000 АК. Следовательно, существует большое разнообразие возможных белковых последовательностей. Линейные цепи сворачиваются в специфические трехмерные конформации, которые определяются последовательностью аминокислот и поэтому также чрезвычайно разнообразны — от полностью волокнистых до глобулярных. Ковалентные дисульфидные связи могут быть введены между цистеиновыми остатками, расположенными в непосредственной близости в трехмерном пространстве, что обеспечивает жесткость полученной трехмерной структуры.Ленточные диаграммы, подобные показанной здесь, являются распространенным способом визуализации белков.
Белковые структуры могут быть определены на атомном уровне с помощью рентгеновской дифракции и нейтронной дифракции кристаллизованных белков, а в последнее время — с помощью спектроскопии ядерного магнитного резонанса (ЯМР) белков в растворе. Однако структура многих белков остается неясной.
Чтобы просмотреть пример третичной структуры в KiNG, нажмите здесь. Это рибонуклеаза А, фермент, ответственный за расщепление РНК.На изображении изображены все атомы одной половины молекулы (голубой для боковых цепей, коричневый для атомов водорода) и только основная цепь и боковые цепи для другой половины. Альтернативный вид показывает атомы основной цепи и Н-связи (фиолетовые). Нажмите «Анимировать», чтобы переключаться между представлениями.
Хотя атомы водорода составляют около половины атомов в белке, они редко проявляются в явном виде, потому что их трудно обнаружить с помощью рентгеновской кристаллографии (из-за низкой электронной плотности) и они очень усложняют картину.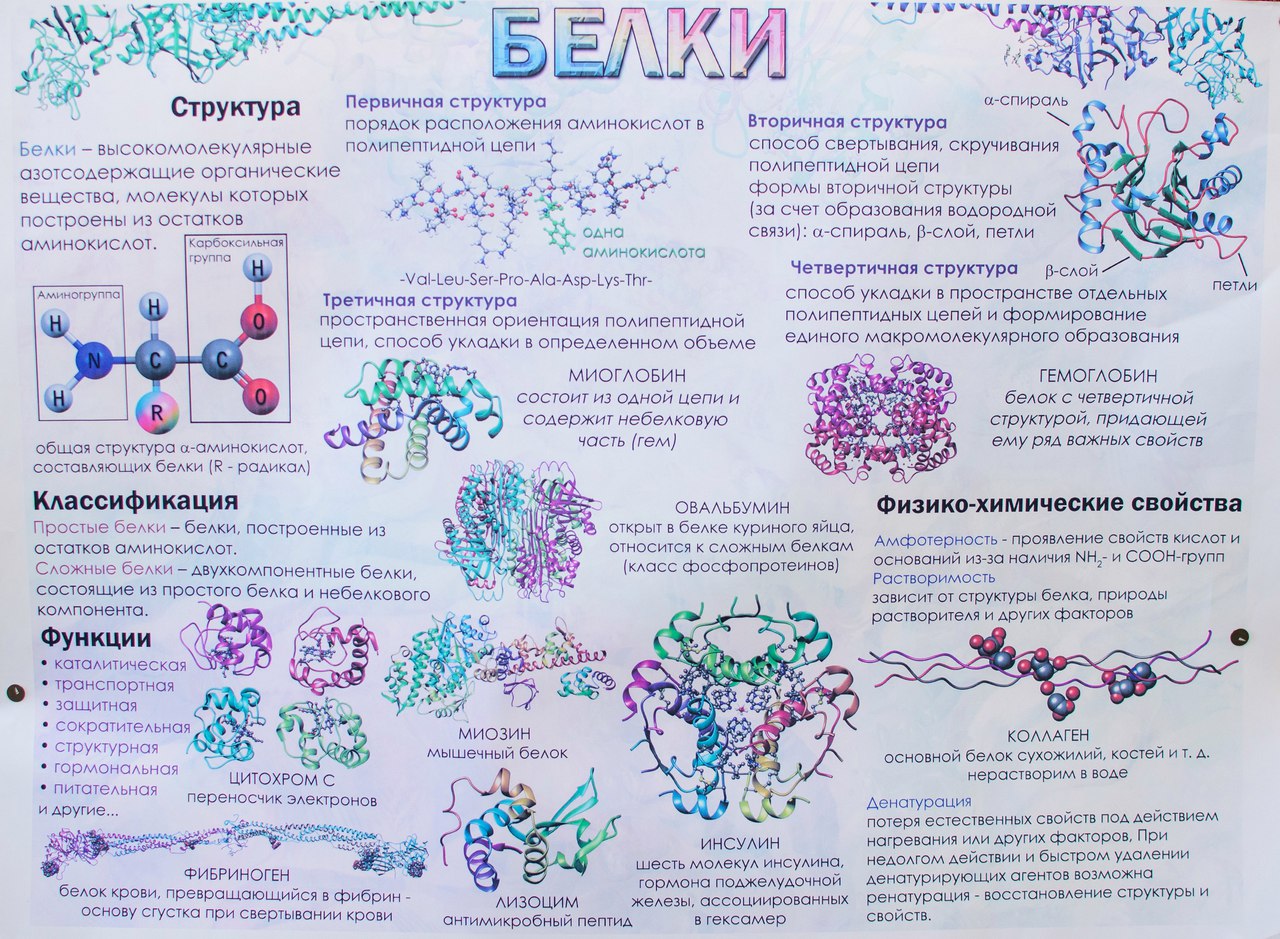 Это изображение рибонуклеазы представляет собой совместную структуру рентгеновской и нейтронной дифракции, в которую всегда включены атомы водорода. Даже без атомов H представление всего атома слишком переполнено, чтобы быть очень полезным, но это хороший способ понять, с чего начинаются упрощенные версии.
Это изображение рибонуклеазы представляет собой совместную структуру рентгеновской и нейтронной дифракции, в которую всегда включены атомы водорода. Даже без атомов H представление всего атома слишком переполнено, чтобы быть очень полезным, но это хороший способ понять, с чего начинаются упрощенные версии.
Некоторый текст адаптирован из : Приложение Kinemage к Branden & Tooze «Введение в структуру белка», глава 2 — МОТИВЫ СТРУКТУРЫ БЕЛКА, Джейн С. и Дэвид С.Ричардсон.
Сворачивание белка — это физический процесс, при котором линейный полипептид сворачивается в свою характерную и функциональную трехмерную структуру. Сворачивание полипептидной цепи сильно зависит от растворимости R-групп AA в воде. Каждый белок существует в виде развернутого полипептида или случайного клубка при трансляции последовательности мРНК в линейную цепь аминокислот. У этого полипептида отсутствует какая-либо стабильная (длительная) трехмерная структура (левая часть соседнего рисунка).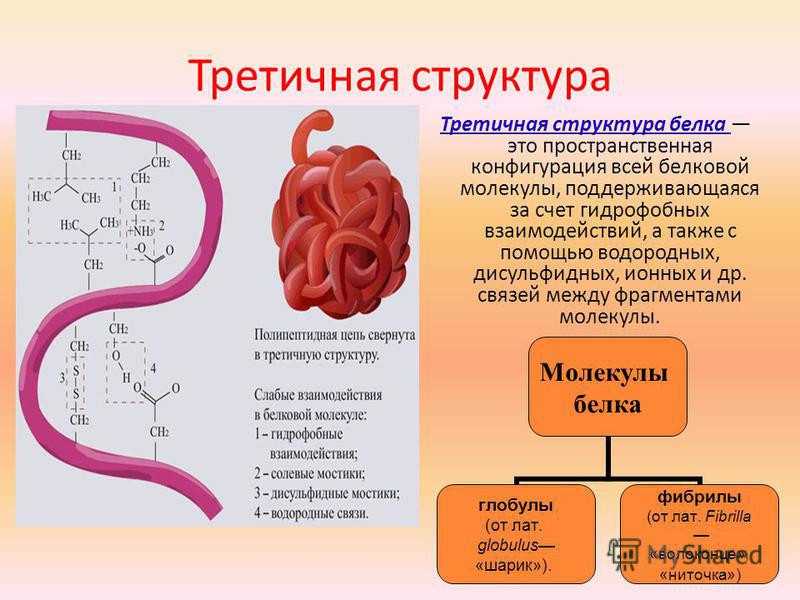 Аминокислоты взаимодействуют друг с другом, образуя четко определенную трехмерную структуру, свернутый белок (правая часть рисунка), известный как нативное состояние. Таким образом, вся информация о нативной укладке содержится в первичной структуре (за это Анфинсен получил Нобелевскую премию), а белки являются самосворачивающимися (хотя in vivo фолдингу полипептидов часто помогают дополнительные молекулы, известные как молекулярные шапероны). ).
Аминокислоты взаимодействуют друг с другом, образуя четко определенную трехмерную структуру, свернутый белок (правая часть рисунка), известный как нативное состояние. Таким образом, вся информация о нативной укладке содержится в первичной структуре (за это Анфинсен получил Нобелевскую премию), а белки являются самосворачивающимися (хотя in vivo фолдингу полипептидов часто помогают дополнительные молекулы, известные как молекулярные шапероны). ).
Минимизация количества гидрофобных боковых цепей, подвергающихся воздействию воды (гидрофобный эффект), является важной движущей силой процесса складывания.Внутримолекулярные водородные связи также способствуют стабильности белка (вспомните об их важности во вторичных структурах). Ионные взаимодействия (притяжение между разноименными электрическими зарядами ионизированных R-групп) также способствуют устойчивости третичных структур. Дисульфидные мостики (ковалентные связи) между соседними остатками цистеина также могут стабилизировать трехмерные структуры.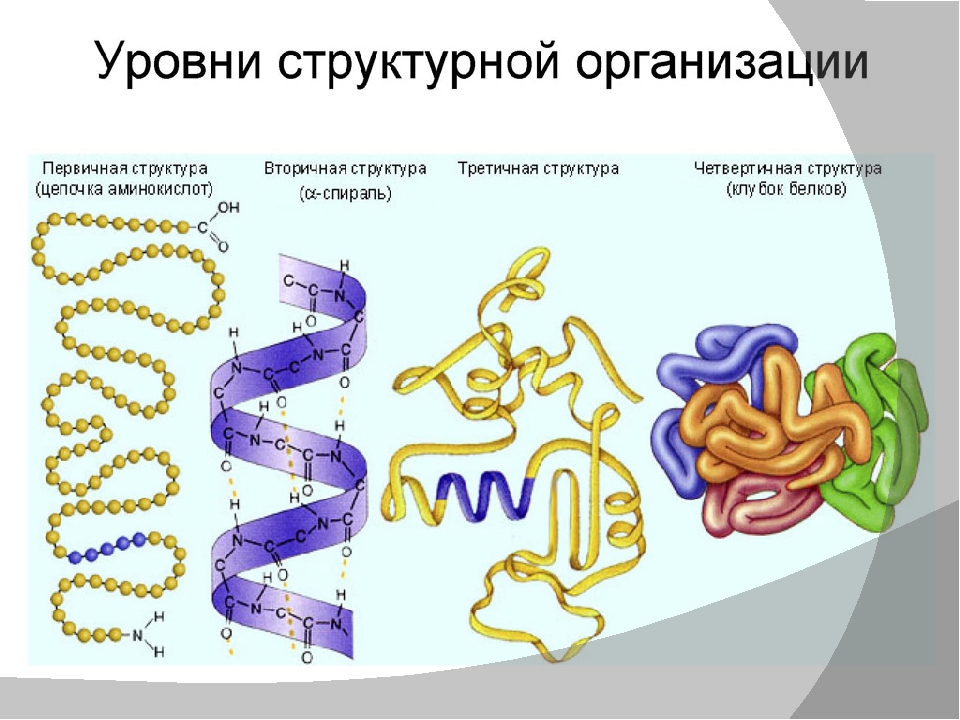 Обратите внимание, что дисульфидные связи редко наблюдаются во внутриклеточных белках из-за восстановительной внутриклеточной среды.
Обратите внимание, что дисульфидные связи редко наблюдаются во внутриклеточных белках из-за восстановительной внутриклеточной среды.
Правильная трехмерная структура белка необходима для его функционирования, хотя некоторые части функциональных белков могут оставаться развернутыми. Неспособность свернуться в нативную структуру обычно приводит к образованию неактивных белков, но в некоторых случаях неправильно свернутые белки имеют модифицированную или токсическую функциональность (например, прионы и амилоидные фибриллы). В соответствии с их функциональной важностью трехмерные структуры белков более консервативны во время эволюции, чем первичные аминокислотные последовательности.
Тем, кто хочет внести свой вклад в науку, играя в игры, я предлагаю вам попробовать FoldIt. Недавно участники этой игры смогли правильно предсказать структуру ретровирусной протеазы. Для тех, кто хочет, чтобы их свободные циклы ЦП использовались с пользой, я предлагаю вам проверить Folding@home.
Четвертичная структура в белках представляет собой наиболее сложную степень организации, до сих пор считающуюся одной молекулой. Чтобы считаться имеющим четвертичную структуру, белок должен иметь две или более пептидных цепей, образующих субъединицы.Субъединицы могут быть разными или одинаковыми и в большинстве случаев расположены симметрично. Обычно белок с двумя субъединицами называется димером; один с тремя субъединицами в тримере; и один с четырьмя субъединицами тетрамера.
Изменения в четвертичной структуре могут происходить за счет конформационных изменений внутри отдельных субъединиц или за счет переориентации субъединиц относительно друг друга. Именно благодаря таким изменениям, лежащим в основе кооперативности и аллостерии в «мультимерных» ферментах, многие белки подвергаются регуляции и выполняют свою физиологическую функцию.Хорошим примером может служить ДНК-полимераза (см. изображение) и ионные каналы. Субъединицы удерживаются вместе теми же типами взаимодействий, которые стабилизируют третичную структуру белков.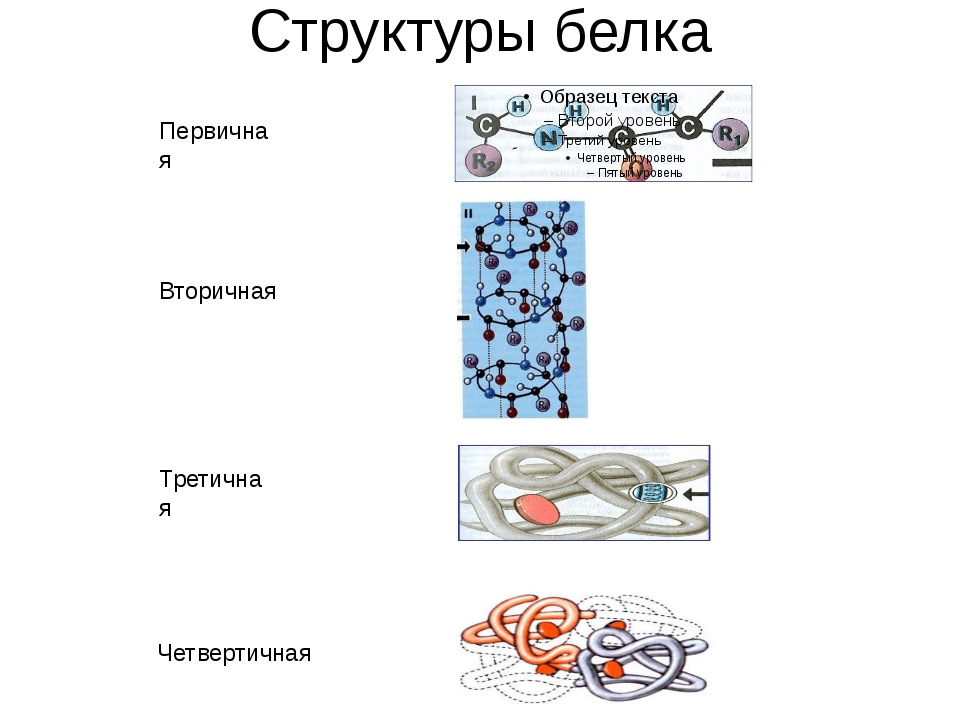
Ведутся споры о том, следует ли определять четвертичную структуру как включающую пептиды, связанные ковалентными (дисульфидными) связями. В CMB мы будем использовать четвертичную структуру для обозначения только расположения субъединиц, которые не связаны ковалентно, хотя внутри отдельных субъединиц могут возникать ковалентные дисульфидные связи.
ProteinTools: набор инструментов для анализа белковых структур | Исследование нуклеиновых кислот
Аннотация
Экспериментальная характеристика и компьютерное предсказание белковых структур становится все более быстрым и точным. Однако анализ белковых структур часто требует от исследователей использования нескольких программных пакетов или веб-серверов, что усложняет задачу. Чтобы обеспечить давно зарекомендовавшие себя структурные анализы в современном, простом в использовании интерфейсе, мы внедрили ProteinTools, набор инструментов веб-сервера для анализа структуры белков. На данный момент ProteinTools объединяет четыре приложения, а именно идентификацию гидрофобных кластеров, сетей водородных связей, солевых мостиков и карт контактов. Во всех случаях входные данные представляют собой идентификатор PDB или загруженную структуру, а выходные данные представляют собой интерактивный динамический веб-интерфейс. Благодаря модульной природе ProteinTools добавление новых приложений станет легкой задачей. Учитывая текущую потребность иметь эти инструменты в едином, быстром и интерпретируемом интерфейсе, мы считаем, что ProteinTools станет важным набором инструментов для более широкого сообщества исследователей белков.Веб-сервер доступен по адресу https://proteintools.uni-bayreuth.de.
На данный момент ProteinTools объединяет четыре приложения, а именно идентификацию гидрофобных кластеров, сетей водородных связей, солевых мостиков и карт контактов. Во всех случаях входные данные представляют собой идентификатор PDB или загруженную структуру, а выходные данные представляют собой интерактивный динамический веб-интерфейс. Благодаря модульной природе ProteinTools добавление новых приложений станет легкой задачей. Учитывая текущую потребность иметь эти инструменты в едином, быстром и интерпретируемом интерфейсе, мы считаем, что ProteinTools станет важным набором инструментов для более широкого сообщества исследователей белков.Веб-сервер доступен по адресу https://proteintools.uni-bayreuth.de.
Graphical Abstract
ProteinTools предлагает анализ гидрофобных кластеров, водородных связей, солевых мостиков и карт контактов в белковых структурах в удобном современном интерфейсе.
Graphical Abstract
ProteinTools предлагает анализ гидрофобных кластеров, водородных связей, солевых мостиков и карт контактов в белковых структурах в простом в использовании современном интерфейсе.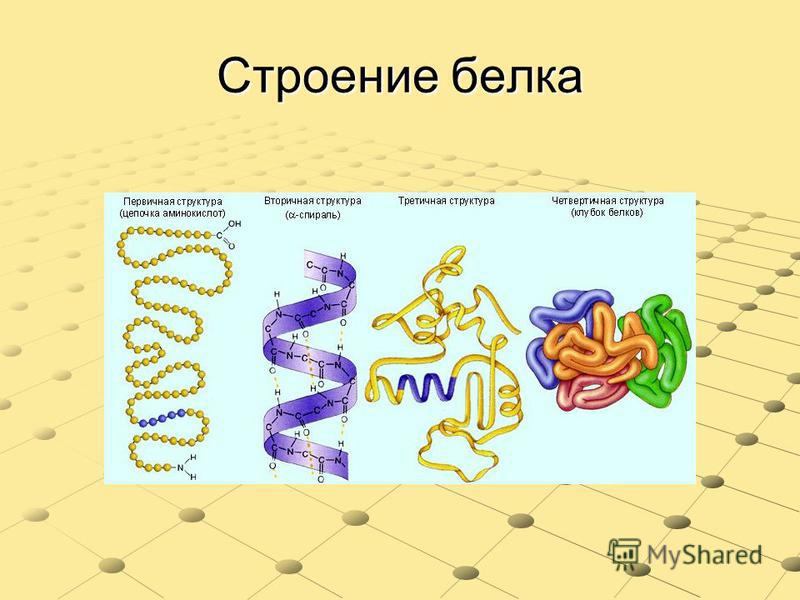
ВВЕДЕНИЕ
Количество депонированных структур в базе данных белков растет с экспоненциальной скоростью, при этом 90% сегодняшних доступных структур были депонированы за последние 20 лет. Мало того, что экспериментальные методы предоставляют множество структурных данных быстрее, чем когда-либо прежде, но и вычислительные усилия по прогнозированию структур значительно продвинулись за последнее десятилетие. Особенно многообещающими оказались недавние методы структурного прогнозирования, основанные на глубоком обучении, такие как DMPfold (1) или AlphaFold (2).Усилия, как в экспериментальных, так и в вычислительных областях, позволили охарактеризовать белковые структуры с беспрецедентной скоростью и детализацией. Поэтому крайне важно, чтобы мы внедрили вычислительные инструменты для анализа этих структур с сопоставимой скоростью и сделали такие инструменты доступными для широкого сообщества.
Трехмерная структура белка важна для его биологической функции, поэтому ее характеристика или точное предсказание имеют жизненно важное значение. Белки складываются в свои нативные структуры в результате взаимодействия, обусловленного различными нековалентными взаимодействиями, такими как водородные связи, силы Ван-дер-Ваальса, гидрофобные и ионные взаимодействия.Таким образом, чтобы понять особенности и функции белка на молекулярном уровне, важно охарактеризовать эти взаимодействия. В то время как большая часть вычислительных усилий в структурной биологии была сосредоточена на реализации инструментов, предсказывающих структуры белков, также было выпущено несколько замечательных инструментов для структурного анализа.
Белки складываются в свои нативные структуры в результате взаимодействия, обусловленного различными нековалентными взаимодействиями, такими как водородные связи, силы Ван-дер-Ваальса, гидрофобные и ионные взаимодействия.Таким образом, чтобы понять особенности и функции белка на молекулярном уровне, важно охарактеризовать эти взаимодействия. В то время как большая часть вычислительных усилий в структурной биологии была сосредоточена на реализации инструментов, предсказывающих структуры белков, также было выпущено несколько замечательных инструментов для структурного анализа.
Многие из этих инструментов возникли из-за необходимости понять взаимодействия в контексте динамики белков (3) и, таким образом, сосредоточиться на анализе молекулярно-динамических (МД) траекторий или являются расширениями наборов инструментов МД.Стоит упомянуть пакеты Gromacs (4) и MDtraj (5), которые позволяют анализировать временную эволюцию молекулярных взаимодействий в языках командной строки и Python. Другие автономные пакеты сосредоточены на анализе этих взаимодействий. В частности, большое внимание привлек анализ водородных связей, поскольку они играют важную роль в фолдинге, структуре и функционировании белков (6). Доступно множество инструментов, которые идентифицируют и анализируют водородные связи. Среди прочего, HBPredicT предполагает наличие водородных связей между водой, лигандами и белками (7).Программы молекулярной визуализации Chimera (8), PyMOL (9) и VMD (10) предлагают несколько инструментов для определения водородных связей в белках и лигандах. Также недавно был разработан алгоритм построения водородных связей в глобальном контексте, HBplot (11).
Другие автономные пакеты сосредоточены на анализе этих взаимодействий. В частности, большое внимание привлек анализ водородных связей, поскольку они играют важную роль в фолдинге, структуре и функционировании белков (6). Доступно множество инструментов, которые идентифицируют и анализируют водородные связи. Среди прочего, HBPredicT предполагает наличие водородных связей между водой, лигандами и белками (7).Программы молекулярной визуализации Chimera (8), PyMOL (9) и VMD (10) предлагают несколько инструментов для определения водородных связей в белках и лигандах. Также недавно был разработан алгоритм построения водородных связей в глобальном контексте, HBplot (11).
Что касается функции белка, то еще одним свойством, которое интересно охарактеризовать, является обнаружение и анализ полостей и каналов. Были разработаны такие инструменты, как PASS (12) и PocketPicker (13), которые обнаруживают карманы связывания, и CAVER (14), веб-сервер для визуализации каталитических карманов в белках. Многие другие инструменты сосредоточены на оценке солевых мостов, например, веб-сервер ESBRI (15) или SBION (16), программа для вычисления солевых мостов из нескольких файлов структуры.
Многие другие инструменты сосредоточены на оценке солевых мостов, например, веб-сервер ESBRI (15) или SBION (16), программа для вычисления солевых мостов из нескольких файлов структуры.
Несмотря на эти значительные достижения, большинство этих инструментов предлагают анализ отдельных структурных свойств и часто доступны в пакетах программного обеспечения, написанных на разных языках программирования. Поэтому пользователям необходимо загрузить и установить несколько инструментов, а также ознакомиться с различной документацией. Таким образом, жизненно важно улучшить такие инструменты, чтобы сделать их интуитивно понятными.Особенно ценны наборы инструментов, которые объединяют множество интересных инструментов на одном веб-сайте, сокращая время анализа пользователей и время обучения. Насколько нам известно, опубликовано не так много веб-серверов в области белков, которые собирают несколько инструментов на одном сайте, хотя мы ожидаем, что эта тенденция изменится. Мы хотели бы выделить инструментарий биоинформатики (17) для анализа белковых последовательностей: он включает, среди прочего, удаленное обнаружение гомологии, предсказание структуры, выравнивание последовательностей и кластеризацию последовательностей. Набор инструментов PlayMolecule (18), с другой стороны, предлагает анализ связывания лигандов, включая такие инструменты, как параметризация лиганда или предсказание аффинности связывания. Кроме того, были собраны наборы инструментов для проверки качества моделей, особенно рентгеновских и ЯМР-структур (19,20). Другими прошлыми инициативами по внедрению наборов инструментов были набор инструментов bPE (21), набор инструментов для инженерии и дизайна белков, и StrucTools, который содержал несколько инструментов, таких как расчет графиков Рамачандрана или расчеты поверхности и объема.Эти последние два примера, к сожалению, больше не поддерживаются.
Набор инструментов PlayMolecule (18), с другой стороны, предлагает анализ связывания лигандов, включая такие инструменты, как параметризация лиганда или предсказание аффинности связывания. Кроме того, были собраны наборы инструментов для проверки качества моделей, особенно рентгеновских и ЯМР-структур (19,20). Другими прошлыми инициативами по внедрению наборов инструментов были набор инструментов bPE (21), набор инструментов для инженерии и дизайна белков, и StrucTools, который содержал несколько инструментов, таких как расчет графиков Рамачандрана или расчеты поверхности и объема.Эти последние два примера, к сожалению, больше не поддерживаются.
В связи с возросшей потребностью в инструментах для быстрого и автономного анализа растущего количества структурных данных мы разработали ProteinTools (https://proteintools.uni-bayreuth.de), набор инструментов для анализа белковых структур. На этом этапе мы добавили четыре приложения: идентификация гидрофобных кластеров, сетей водородных связей, солевых мостиков и карт контактов. Гидрофобные кластеры препятствуют проникновению молекул воды в ядро белка и служат телами стабильности в высокоэнергетических частично свернутых состояниях.Предыдущее программное обеспечение и серверы для расчета гидрофобных кластеров, такие как алгоритм «Контакты структурных единиц» (CSU) (22) и веб-сервер BASIC (23), к сожалению, больше не доступны. С недавним появлением мощных веб-инструментов молекулярной визуализации, отличных от Adobe Flash/Java, таких как веб-приложение Mol* (https://molstar.org/), мы можем вернуть сообществу вычисление гидрофобных кластеров. Сети водородных связей обеспечивают связь между остатками, удаленными друг от друга в структуре белка (6,24).Они помогают стабилизировать белок и играют роль в аллостерии. Несмотря на их относительно простую идентификацию, веб-инструмент, который анализирует и отображает сети водородных связей, все еще отсутствует. Другими часто запрашиваемыми исследователями белков инструментами анализа являются вычисление солевых мостиков и карт контактов.
Гидрофобные кластеры препятствуют проникновению молекул воды в ядро белка и служат телами стабильности в высокоэнергетических частично свернутых состояниях.Предыдущее программное обеспечение и серверы для расчета гидрофобных кластеров, такие как алгоритм «Контакты структурных единиц» (CSU) (22) и веб-сервер BASIC (23), к сожалению, больше не доступны. С недавним появлением мощных веб-инструментов молекулярной визуализации, отличных от Adobe Flash/Java, таких как веб-приложение Mol* (https://molstar.org/), мы можем вернуть сообществу вычисление гидрофобных кластеров. Сети водородных связей обеспечивают связь между остатками, удаленными друг от друга в структуре белка (6,24).Они помогают стабилизировать белок и играют роль в аллостерии. Несмотря на их относительно простую идентификацию, веб-инструмент, который анализирует и отображает сети водородных связей, все еще отсутствует. Другими часто запрашиваемыми исследователями белков инструментами анализа являются вычисление солевых мостиков и карт контактов. Поэтому мы также включили решения этих проблем в ProteinTools. Чтобы продемонстрировать применение этих четырех инструментов, мы используем домен Di-III_14 в качестве примера, разработанную IF-3-подобную укладку с 74 аминокислотами, которая обладает необычными свойствами укладки (25,26).
Поэтому мы также включили решения этих проблем в ProteinTools. Чтобы продемонстрировать применение этих четырех инструментов, мы используем домен Di-III_14 в качестве примера, разработанную IF-3-подобную укладку с 74 аминокислотами, которая обладает необычными свойствами укладки (25,26).
МАТЕРИАЛЫ И МЕТОДЫ
Гидрофобные кластеры
Было высказано предположение, что боковые цепи остатков изолейцина (ILE), лейцина (LEU) и валина (VAL) часто образуют гидрофобные или так называемые (ILV) кластеры, которые предотвращают проникновение молекул воды и служат ядрами стабильности при высоких -энергетические частично свернутые состояния (23). Различные инструменты для анализа гидрофобных кластеров исключительно из белковых последовательностей были разработаны (27) и недавно стали доступны в виде пакета Python (28).Другая возможность заключается в идентификации гидрофобных кластеров непосредственно в структуре белка. Их вычисление основано на алгоритме «Контакты структурных единиц» (CSU), который также широко используется для расчета контактных карт (22, 29). Хотя алгоритм CSU изначально был выпущен в виде пакета и веб-сервера, к сожалению, оба они больше не доступны. Совсем недавно алгоритм CSU был применен к частному случаю вычисления контактов между гидрофобными атомами для определения кластеров ILV, и он был выпущен на веб-сервере BASIC, который также больше недоступен (23).Первоначальный алгоритм работает следующим образом: два атома А и В считаются контактирующими, если молекула растворителя, помещенная на поверхность сферы А, перекрывается со сферой Ван-дер-Ваальса атома В плюс сфера, образованная другой молекулой растворителя (30). . Атомы считаются сферами фиксированного радиуса (31). Если молекула воды проникает через несколько сфер атомов в любом положении, считается, что контакт принадлежит той, центр которой находится ближе всего к центру атома A. во внимание, а затем извлекает координаты соседних атомов.
Их вычисление основано на алгоритме «Контакты структурных единиц» (CSU), который также широко используется для расчета контактных карт (22, 29). Хотя алгоритм CSU изначально был выпущен в виде пакета и веб-сервера, к сожалению, оба они больше не доступны. Совсем недавно алгоритм CSU был применен к частному случаю вычисления контактов между гидрофобными атомами для определения кластеров ILV, и он был выпущен на веб-сервере BASIC, который также больше недоступен (23).Первоначальный алгоритм работает следующим образом: два атома А и В считаются контактирующими, если молекула растворителя, помещенная на поверхность сферы А, перекрывается со сферой Ван-дер-Ваальса атома В плюс сфера, образованная другой молекулой растворителя (30). . Атомы считаются сферами фиксированного радиуса (31). Если молекула воды проникает через несколько сфер атомов в любом положении, считается, что контакт принадлежит той, центр которой находится ближе всего к центру атома A. во внимание, а затем извлекает координаты соседних атомов. В случае альтернативных конформаций рассматривается только первое состояние. Это атомы, которые ближе, чем сумма двух радиусов Ван-дер-Ваальса, каждый из которых увеличен на радиус молекулы воды (1,4 Å). Следовательно, чтобы два атома углерода считались кандидатами на атомные контакты, они должны находиться в пределах 6,56 Å. ProteinTools разделяет каждую атомную сферу на 610 однородных секций, используя сетку Фибоначчи (32,33). Площадь соответствует 0,0016 общей площади сферы. Затем алгоритм оценивает, перекрываются ли какие-либо из 610 секций со своими соседями.Если это так, то контакт сечения объявляется принадлежащим атому, центр которого находится ближе всего к центру исходной сферы.
В случае альтернативных конформаций рассматривается только первое состояние. Это атомы, которые ближе, чем сумма двух радиусов Ван-дер-Ваальса, каждый из которых увеличен на радиус молекулы воды (1,4 Å). Следовательно, чтобы два атома углерода считались кандидатами на атомные контакты, они должны находиться в пределах 6,56 Å. ProteinTools разделяет каждую атомную сферу на 610 однородных секций, используя сетку Фибоначчи (32,33). Площадь соответствует 0,0016 общей площади сферы. Затем алгоритм оценивает, перекрываются ли какие-либо из 610 секций со своими соседями.Если это так, то контакт сечения объявляется принадлежащим атому, центр которого находится ближе всего к центру исходной сферы.
Алгоритм выполняется для всех атомов до тех пор, пока не будет вычислена матрица площадей остатка против остатка. По умолчанию ProteinTools определяет, что два остатка находятся в контакте, когда их общая площадь перекрытия составляет не менее 10 Å 2 . Смежная матрица преобразуется в граф, где каждый компонент соответствует (гидрофобному) кластеру. Общая площадь кластера вычисляется как сумма отдельных областей остатков, которые его составляют.ProteinTools показывает каждый из рассчитанных гидрофобных кластеров разным цветом на интерактивной панели. Свойства каждого кластера сведены в таблицу. Результаты доступны для скачивания в виде сеанса PyMOL (9) и таблицы. Реализация гидрофобных кластеров в ProteinTools основана на пакетах SciPy и NumPy Python.
Общая площадь кластера вычисляется как сумма отдельных областей остатков, которые его составляют.ProteinTools показывает каждый из рассчитанных гидрофобных кластеров разным цветом на интерактивной панели. Свойства каждого кластера сведены в таблицу. Результаты доступны для скачивания в виде сеанса PyMOL (9) и таблицы. Реализация гидрофобных кластеров в ProteinTools основана на пакетах SciPy и NumPy Python.
Сети с водородными связями
Сети водородных связей представляют собой сети водородных связей, которые соединяют боковые цепи нескольких остатков в белке.Чтобы вычислить различные сети водородных связей, ProteinTools сначала протонирует заданные пользователем координаты с помощью PROPKA (34) и PDB2PQR (35). Для воспроизводимости мы (повторно) протонируем все PDB и рассматриваем только первую конформацию альтернативных боковых цепей. Следуя алгоритму PDB2PQR, протоны добавляют после оценки значений pKa для каждого остатка при pH 7,0 (34).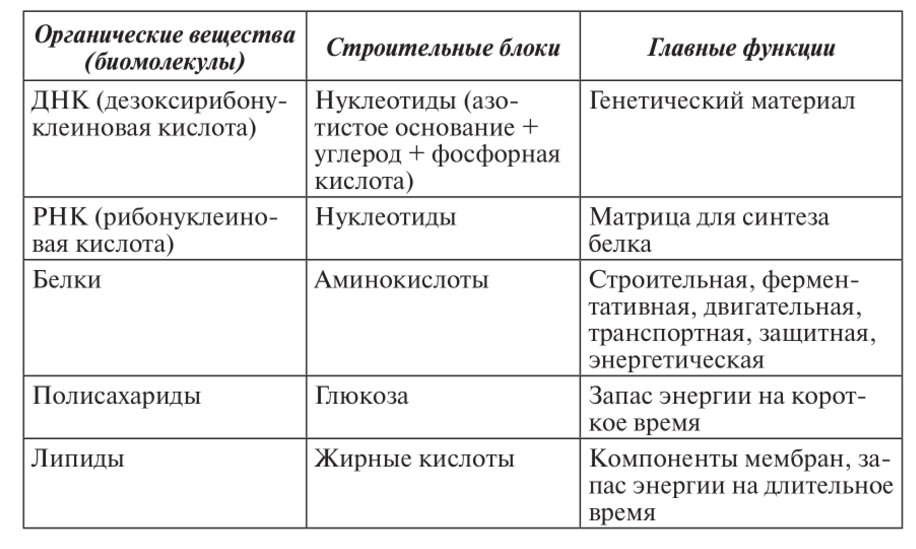 Кроме того, боковые цепи переворачиваются и вращаются для оптимизации локальных сетей водородных связей (35). После протонирования ProteinTools вычисляет все водородные сети в боковых цепях белка, используя алгоритм Бейкера-Хаббарда (36).Мы выбираем отсечки ϑ > 120° и d < 2,5 Å, где ϑ — угол, определяемый тремя атомами, а d — расстояние между донорным атомом водорода и акцепторным атомом. Серверная часть ProteinTools полагается на пакет MDtraj для некоторых из этих вычислений (5). В этом методе рассматриваются атомы «NH» и «OH» в качестве доноров, а также кислорода и азота в качестве акцепторов. Как только все водородные связи вычислены, любые два остатка считаются связанными, если между ними можно найти последовательный путь водородных связей.ProteinTools назначает каждой сети свой цвет на интерактивной панели Mol*. Каждая водородная связь отдельно описана в таблице. Таблицы и структуры белков можно загрузить в виде файлов CSV и сеансов PyMOL (9) соответственно.
Кроме того, боковые цепи переворачиваются и вращаются для оптимизации локальных сетей водородных связей (35). После протонирования ProteinTools вычисляет все водородные сети в боковых цепях белка, используя алгоритм Бейкера-Хаббарда (36).Мы выбираем отсечки ϑ > 120° и d < 2,5 Å, где ϑ — угол, определяемый тремя атомами, а d — расстояние между донорным атомом водорода и акцепторным атомом. Серверная часть ProteinTools полагается на пакет MDtraj для некоторых из этих вычислений (5). В этом методе рассматриваются атомы «NH» и «OH» в качестве доноров, а также кислорода и азота в качестве акцепторов. Как только все водородные связи вычислены, любые два остатка считаются связанными, если между ними можно найти последовательный путь водородных связей.ProteinTools назначает каждой сети свой цвет на интерактивной панели Mol*. Каждая водородная связь отдельно описана в таблице. Таблицы и структуры белков можно загрузить в виде файлов CSV и сеансов PyMOL (9) соответственно.
Расчеты солевого мостика и распределения заряда
Мы определяем сети солевых мостиков, выбирая все кислые атомы кислорода и все основные атомы азота и вычисляя матрицу их расстояний «все против всех». Те пары с расстояниями менее 4 Å считаются солевыми мостиками.Альтернативное расположение сайдчейнов не рассматривается, во всех случаях сохраняется только первое состояние. ProteinTools отображает каждый кластер солевого мостика отдельно в интерактивном окне. Это приложение также обеспечивает вычисление параметров κ (каппа) и доли заряженных остатков (FCR), которые в основном изучались в лаборатории Паппу (37). κ является мерой степени разделения заряда в последовательности. FCR представляет собой долю заряженных остатков в последовательности. Эти значения можно использовать для прогнозирования компактности белков.ProteinTools вычисляет эти значения с помощью пакета CIDER (38). Белковые структуры с визуализированными солевыми мостиками также можно загрузить в виде сеанса PyMOL (9).
Те пары с расстояниями менее 4 Å считаются солевыми мостиками.Альтернативное расположение сайдчейнов не рассматривается, во всех случаях сохраняется только первое состояние. ProteinTools отображает каждый кластер солевого мостика отдельно в интерактивном окне. Это приложение также обеспечивает вычисление параметров κ (каппа) и доли заряженных остатков (FCR), которые в основном изучались в лаборатории Паппу (37). κ является мерой степени разделения заряда в последовательности. FCR представляет собой долю заряженных остатков в последовательности. Эти значения можно использовать для прогнозирования компактности белков.ProteinTools вычисляет эти значения с помощью пакета CIDER (38). Белковые структуры с визуализированными солевыми мостиками также можно загрузить в виде сеанса PyMOL (9).
Карты контактов
Карты контактов белков представляют собой расстояния между всеми парами аминокислотных остатков в виде матрицы. ProteinTools вычисляет карты контактов, вычисляя матрицу расстояний «все против всех» остатков и беря минимальное расстояние между любыми двумя атомами в двух оцениваемых остатках.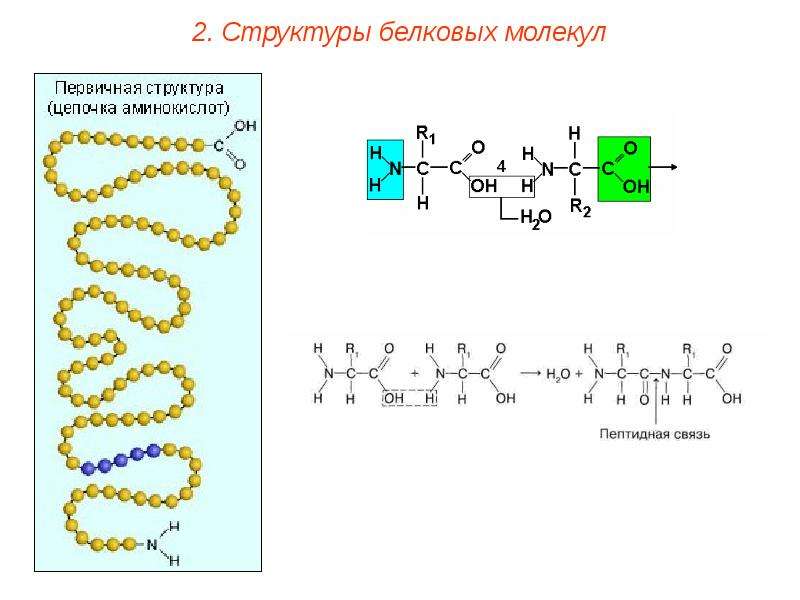 Необработанные данные отображаются на интерактивной панели, которую можно экспортировать в виде таблицы CSV.
Необработанные данные отображаются на интерактивной панели, которую можно экспортировать в виде таблицы CSV.
Внедрение белковых инструментов
ProteinTools разработан с использованием фреймворка Django Python (версия 3.1.2). Бэкенд полностью реализован на Python. Интерфейс веб-сайта разработан с помощью JavaScript с использованием фреймворка Bootstrap (версия 4.2). Белки визуализируются с помощью веб-пакета PDBe Molstar (39). Конкретные пакеты Python, используемые в каждом приложении, указаны в разделах выше.Все приложения требуют код PDB или определяемую пользователем структуру PDB в качестве входных данных и предоставляют интерактивное окно в качестве выходных данных. Данные можно загрузить в виде таблиц CSV, а для внешней визуализации сеансы PyMOL предоставляются, когда это необходимо. Сеть предоставляет свободный доступ всем пользователям и не требует входа в систему. Документация предоставляется для каждого приложения отдельно на сайте https://proteintools. uni-bayreuth.de.
uni-bayreuth.de.
РЕЗУЛЬТАТЫ
Мы демонстрируем четыре приложения ProteinTools на примере белка Di-III_14 (код PDB 2LN3).В 2012 г. в исключительной работе Koga et al. (26), были определены правила проектирования идеализированных белковых структур, и с использованием этих принципов было разработано несколько белковых складок, встречающихся в природе. Белки, разработанные в этой работе, включали укладку, подобную ферредоксину, укладку Россмана 2 × 2 и 3 × 1, укладку P-петли 2 × 2 и IF3-подобную укладку. Один из IF3-подобных складчатых дизайнов, Di-III_14, был дополнительно проанализирован Робертом Мэтьюзом и его лабораторией (25). Di-III_14 представляет собой белок длиной 74 аминокислоты с четырьмя β-нитями и двумя альфа-спиралями, упакованными на одной стороне β-листа.Порядок β-цепей равен 1243, причем 4 из них антипараллельны остальным. Исследователи заметили, что, хотя Di-III_14 разворачивается в двух состояниях в миллисекундной шкале времени, он остается свернутым в течение нескольких секунд при высоких концентрациях мочевины, что является необычным свойством среди природных белков.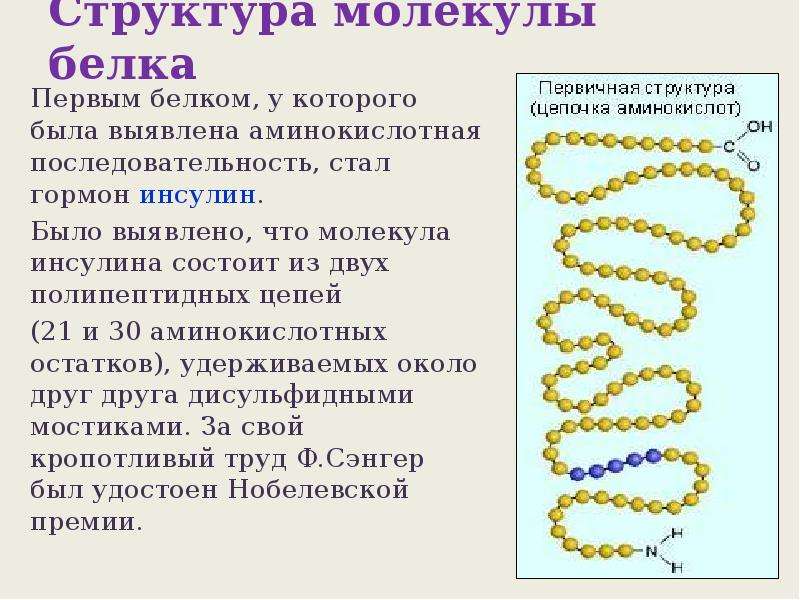 Эксперименты выявили многочисленные высокоэнергетические состояния, которые взаимопревращаются друг с другом в медленных временных масштабах, что структурно соответствовало образованию больших электростатических сетей и гидрофобных кластеров. Здесь мы используем ProteinTools, чтобы показать вычисление этих свойств.
Эксперименты выявили многочисленные высокоэнергетические состояния, которые взаимопревращаются друг с другом в медленных временных масштабах, что структурно соответствовало образованию больших электростатических сетей и гидрофобных кластеров. Здесь мы используем ProteinTools, чтобы показать вычисление этих свойств.
Di-III_14 содержит большой гидрофобный кластер
Басак и др. выполнил ЯМР-анализ водородного обмена (HDX) Di-III_14, который показал процесс обмена между конформационными состояниями в медленных временных масштабах, что резко отличается от быстрого процесса разворачивания, обнаруженного при денатурации хлорида гуанидиния (25). Хотя оба процесса, как правило, дают сопоставимые оценки в природных белках, оптимизация стабильности, проводимая в процессе дизайна белка, может привести к множественным взаимодействиям, которые стабилизируют плотно упакованную внутреннюю часть, что приводит к сложному поведению, не наблюдаемому в природных белках. Авторы нанесли на структуру строго защищенные атомы водорода основной цепи (NH) и обнаружили, что они соответствуют большому гидрофобному ядру, окруженному полярными боковыми цепями. Здесь мы вычислили гидрофобные кластеры Di-III_14, чтобы дополнить эти результаты, анализ, который можно просмотреть по адресу https://proteintools.uni-bayreuth.de/clusters/structure/2ln3 (рис. 1A). Анализ гидрофобных кластеров выявил один кластер, состоящий из 14 остатков, общей площадью 1654,0 Å 2 .Кластер охватывает остатки через все элементы вторичной структуры, причем большинство аминокислот принадлежит β-цепям. Площадь на остаток составляет 44,7 Å 2 , а всего между остатками имеется 37 контактов. Мы задались вопросом, соответствуют ли эти значения особенно плотно упакованному IF3-подобному белку. Мы сравнили гидрофобные кластеры Di-III_14 с кластерами природных IF3-подобных белков. С этой целью мы загрузили все домены из SCOPe (40), базы данных, которая классифицирует белковые структуры в соответствии с их топологией и эволюционными отношениями.
Авторы нанесли на структуру строго защищенные атомы водорода основной цепи (NH) и обнаружили, что они соответствуют большому гидрофобному ядру, окруженному полярными боковыми цепями. Здесь мы вычислили гидрофобные кластеры Di-III_14, чтобы дополнить эти результаты, анализ, который можно просмотреть по адресу https://proteintools.uni-bayreuth.de/clusters/structure/2ln3 (рис. 1A). Анализ гидрофобных кластеров выявил один кластер, состоящий из 14 остатков, общей площадью 1654,0 Å 2 .Кластер охватывает остатки через все элементы вторичной структуры, причем большинство аминокислот принадлежит β-цепям. Площадь на остаток составляет 44,7 Å 2 , а всего между остатками имеется 37 контактов. Мы задались вопросом, соответствуют ли эти значения особенно плотно упакованному IF3-подобному белку. Мы сравнили гидрофобные кластеры Di-III_14 с кластерами природных IF3-подобных белков. С этой целью мы загрузили все домены из SCOPe (40), базы данных, которая классифицирует белковые структуры в соответствии с их топологией и эволюционными отношениями. Идентификатор SCOPe для IF3-подобных белков — d.68. После извлечения всех белков укладки d.68 мы отбросили те, у которых длина последовательности превышает 150 аминокислот, что привело к 43 членам, указанным в дополнительной таблице S1. Визуализация этих структур с помощью ProteinTools выявила среднее число кластеров 2,2 на структуру, причем кластер, расположенный между спиралями и тяжами, был самым большим во всех случаях. Средняя площадь этого кластера среди белков составляет 1957,5 Å 2 , что немного больше, чем у Di-III_14 (1654.0 Å 2 ), но в пределах одного стандартного отклонения (±1078,9 Å 2 ). Среднее число остатков составляет 14,1, что соответствует результатам для Di-III_14. Репрезентативный IF3-подобный белок с тремя кластерами и площадью 1978 Å 2 для самого большого кластера показан на рисунке 1b для сравнения. В свете этих результатов мы не можем заключить, что гидрофобный кластер Di-III_14 значительно отличается от такового в IF3-подобных природных белках.
Идентификатор SCOPe для IF3-подобных белков — d.68. После извлечения всех белков укладки d.68 мы отбросили те, у которых длина последовательности превышает 150 аминокислот, что привело к 43 членам, указанным в дополнительной таблице S1. Визуализация этих структур с помощью ProteinTools выявила среднее число кластеров 2,2 на структуру, причем кластер, расположенный между спиралями и тяжами, был самым большим во всех случаях. Средняя площадь этого кластера среди белков составляет 1957,5 Å 2 , что немного больше, чем у Di-III_14 (1654.0 Å 2 ), но в пределах одного стандартного отклонения (±1078,9 Å 2 ). Среднее число остатков составляет 14,1, что соответствует результатам для Di-III_14. Репрезентативный IF3-подобный белок с тремя кластерами и площадью 1978 Å 2 для самого большого кластера показан на рисунке 1b для сравнения. В свете этих результатов мы не можем заключить, что гидрофобный кластер Di-III_14 значительно отличается от такового в IF3-подобных природных белках.
Рис. 1.
Рис. 1.
Сети водородных связей
Басак и др. наблюдал две электростатические сети, одна из которых охватывает поверхности α1 и α2, а другая содержит квартет солевых мостиков, которые связывают две внутренние β-цепи, β2 и β4. Чтобы резюмировать эти результаты, мы вычислили сети водородных связей Di-III_14 (https://proteintools.uni-bayreth.de/bonds/structure/2ln3). ProteinTools вычисляет сети водородных связей между боковыми цепями, рассматривая доноры и акцепторы азота и кислорода в пределах 2.5 Å и угол более 120° (см. Материалы и методы). Di-III_14 содержит восемь сетей водородных связей (рис. 2). Самый крупный из них, аналогичный описанию Basak et al. охватывает β1, β2 и β4 и содержит шесть остатков (сеть водородных связей 4, рисунок 2, светло-зеленый). Остатки представляют собой Thr6, Glu30, Glu32, Gln64, Arg69 и Arg71. Еще две сети усиливают взаимодействие внутренних нитей: сеть 5 (синяя, Asp34 и Lys67) и сеть 7 (темно-желтая, Asp28 и Ser 75).
Рис. 2.
Рисунок 2.
В нашем анализе мы наблюдаем в общей сложности четыре сети водородных связей в спиралях, причем три из них в основном охватывают α2. Сеть 3 (оранжевая) включает остатки Glu 45, Glu49 и Lys61 и объединяет α2 и β3. Точно так же сеть 0 (красный) связывает нити и спирали, объединяя в сеть Ser9 в β1 с Asn11 и Glu14 в α1. Две другие сети соответствуют сети 2 (желтая), полностью содержащейся в α2 (остатки Asp50 и Lys 54) и сети 1 (темно-зеленая), связывающей α1 и α2 через Lys12 и Glu43.
Соляные мосты
Приложение ProteinTool для создания солевых мостиков позволяет находить сети солевых мостиков в белке и вычислять параметры разделения заряда (37). Мы рассчитали солевые мостики Di-III_14 на https://proteintools.uni-bayreuth.de/salt/structure/2ln3 (рис. 3). Di-III_14 имеет шесть сетей солевых мостов. Самый большой из них, солевой мостик 4 (выделен), включает многие остатки в сети водородных связей 4: Glu30, Glu32, Arg69 и Arg71, и вместе с солевым мостиком 3 (Lys67 и Asp34) охватывает внутренний β-лист. Солевой мостик 2 связывает элементы β4 и α2 (Lys61, Glu45 и Glu49), тогда как солевой мостик 0 связывает α1 и α2 (Lys12, Glu13, Glu40 и Glu43). Наконец, солевой мостик 1 с остатками Asp50, Lys53 и Lys54 охватывает половину α2. Наши сети согласны с Basak et al. , с некоторыми отличиями, возникающими из-за нашего более строгого ограничения расстояния 4 Å между парами остатков.
Солевой мостик 2 связывает элементы β4 и α2 (Lys61, Glu45 и Glu49), тогда как солевой мостик 0 связывает α1 и α2 (Lys12, Glu13, Glu40 и Glu43). Наконец, солевой мостик 1 с остатками Asp50, Lys53 и Lys54 охватывает половину α2. Наши сети согласны с Basak et al. , с некоторыми отличиями, возникающими из-за нашего более строгого ограничения расстояния 4 Å между парами остатков.
Рисунок 3.
Рисунок 3.
Basak et al. предположил, что необычно большой состав заряженных боковых цепей отличает механизм сворачивания DI-III_14 от природных белков.Авторы построили график доли заряженных остатков (FCR) в зависимости от κ почти для всего протеома термофила Sulfolobus solfataricus и обнаружили, что Di-III_14 появляется в другой области, чем остальные белки. ProteinTools также может вычислять эти параметры, давая FCR 0,35 и κ 0,25, в соответствии с Basak et al. Результаты . Мы задались вопросом, распространяются ли эти различия между Di-III_14 и природными белками на другие конструкции в работе Koga et al. (21). С этой целью мы взяли все последовательности белка SCOPe из соответствующих складок в Koga et al. и сравнил их с дизайном. Спроектированные складки и их идентификаторы SCOPe: Складка-I: ферредоксиноподобная складка (d.58), Складка-II: Россманн 2 × 2 (c.2), Складка-III: IF3-подобная складка (d.68) , складка IV: П-петля 2×2 сложения (с.37), Складка V: Россманна 3×1 (с.23). Природные белки, принадлежащие к этим складкам, имеют тенденцию иметь значения FCR около 0,25 и значения κ около 0,2, кластеризуясь в аналогичной области пространства (дополнительный рисунок S1a).Однако разработанные белки, как правило, имеют более высокие значения FCR (FCR ≥ 0,35 в 4/5 случаях) и более низкие значения κ (κ = 0,12–0,14 в 4/5 случаях) и, следовательно, появляются на периферии (дополнительный рисунок S1b). Этот эффект может быть связан с чрезмерной стабилизацией за счет введения взаимодействий в процессе проектирования белков для обеспечения стабильности конструкций, но эта гипотеза требует дальнейшего изучения.
(21). С этой целью мы взяли все последовательности белка SCOPe из соответствующих складок в Koga et al. и сравнил их с дизайном. Спроектированные складки и их идентификаторы SCOPe: Складка-I: ферредоксиноподобная складка (d.58), Складка-II: Россманн 2 × 2 (c.2), Складка-III: IF3-подобная складка (d.68) , складка IV: П-петля 2×2 сложения (с.37), Складка V: Россманна 3×1 (с.23). Природные белки, принадлежащие к этим складкам, имеют тенденцию иметь значения FCR около 0,25 и значения κ около 0,2, кластеризуясь в аналогичной области пространства (дополнительный рисунок S1a).Однако разработанные белки, как правило, имеют более высокие значения FCR (FCR ≥ 0,35 в 4/5 случаях) и более низкие значения κ (κ = 0,12–0,14 в 4/5 случаях) и, следовательно, появляются на периферии (дополнительный рисунок S1b). Этот эффект может быть связан с чрезмерной стабилизацией за счет введения взаимодействий в процессе проектирования белков для обеспечения стабильности конструкций, но эта гипотеза требует дальнейшего изучения.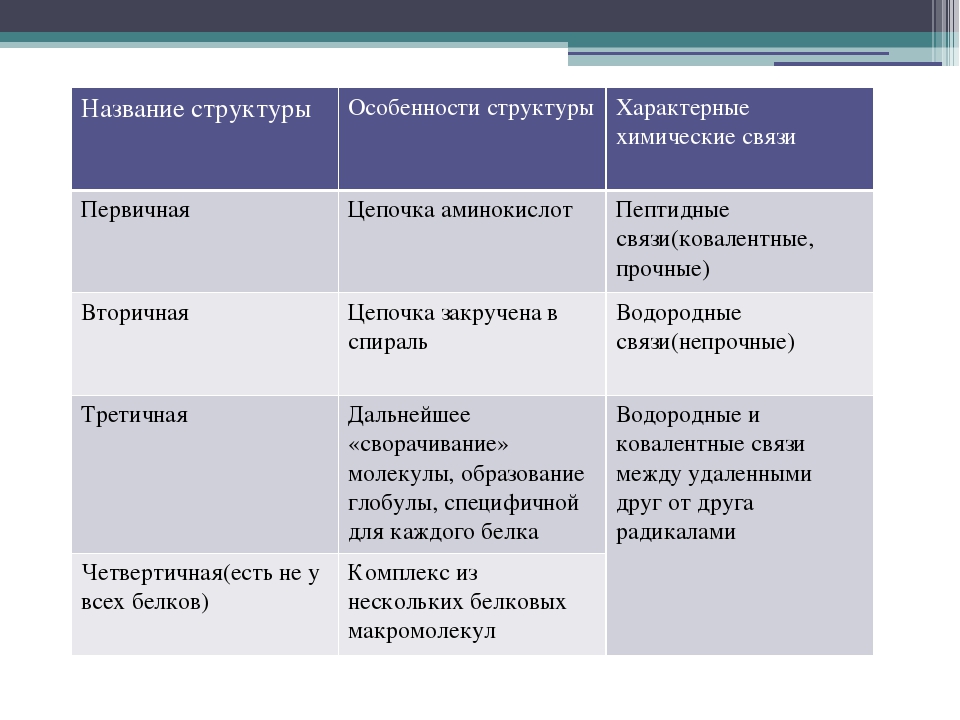
Карты контактов
Карты контактов белков представляют собой расстояние между всеми возможными парами аминокислот и обеспечивают сокращенное представление белковых структур, инвариантных к вращениям и трансляциям.Они широко используются в методах машинного обучения и могут применяться для реконструкции трехмерных структур (41) или анализа сходства белков (42). Поэтому быстрое вычисление карт контактов полезно для самых разных целей. В качестве примера мы рассчитали карту контактов Di-III_14 (рис. 4).
Рисунок 4.
Рисунок 4.
ОБСУЖДЕНИЕ
В то время как новые методы и автоматизация процессов произвели революцию в получении структурных данных белков, существует большая потребность в адаптации инструментов для их анализа.Веб-приложения стали особенно полезными в последние годы: они (i) не требуют установки, (ii) доступны с любого компьютера, подключенного к Интернету, и (iii) освобождают пользователя от изучения определенных программ.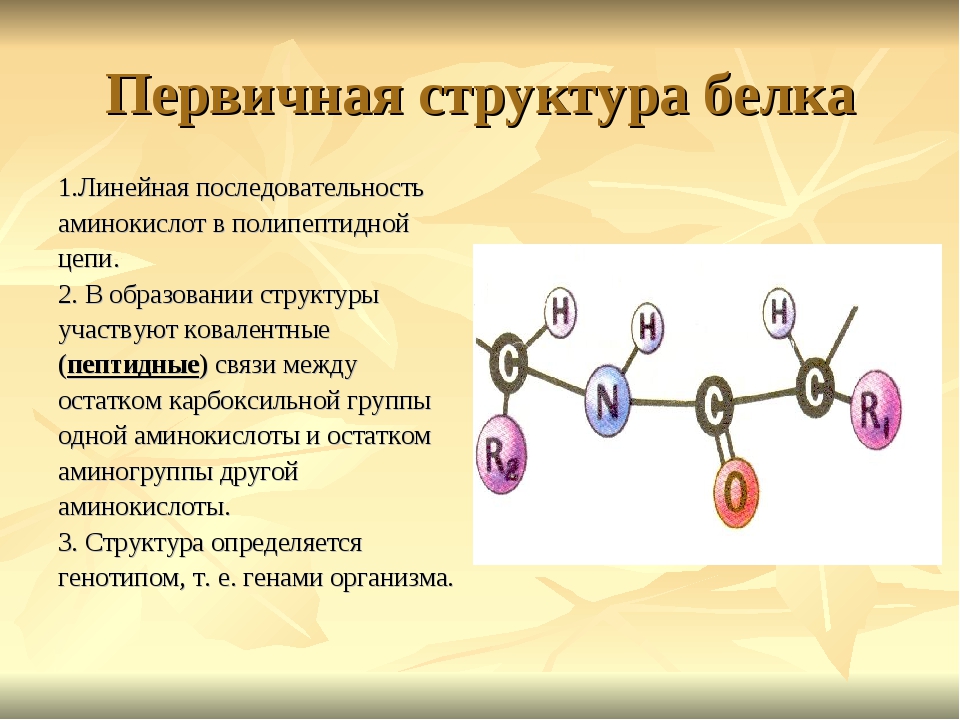 Среди веб-серверов наборы инструментов особенно ценны, поскольку они собирают несколько приложений, для которых в противном случае потребовались бы различные пакеты или веб-серверы. Эти наборы инструментов не только упрощают использование, но также помогают направлять анализ и более полно просматривать белковые структуры и выявлять общие закономерности (43).Руководствуясь этими текущими потребностями, мы внедрили ProteinTools в виде модульного инструментария для анализа белковых структур. На данный момент мы внедрили четыре столь необходимых инструмента анализа: гидрофобные кластеры, сети водородных связей, солевые мосты и карты контактов. Его выпуск особенно своевременен и полезен для сообщества, учитывая, что, насколько нам известно, в настоящее время нет другого веб-сервера для расчета гидрофобных кластеров и сетей водородных связей. Модульный характер набора инструментов упрощает добавление других приложений в ProteinTools.Мы предполагаем интеграцию приложения для генерации мутантов и оценки их ΔΔG°, а также вычисления полостей в ближайшем будущем.
Среди веб-серверов наборы инструментов особенно ценны, поскольку они собирают несколько приложений, для которых в противном случае потребовались бы различные пакеты или веб-серверы. Эти наборы инструментов не только упрощают использование, но также помогают направлять анализ и более полно просматривать белковые структуры и выявлять общие закономерности (43).Руководствуясь этими текущими потребностями, мы внедрили ProteinTools в виде модульного инструментария для анализа белковых структур. На данный момент мы внедрили четыре столь необходимых инструмента анализа: гидрофобные кластеры, сети водородных связей, солевые мосты и карты контактов. Его выпуск особенно своевременен и полезен для сообщества, учитывая, что, насколько нам известно, в настоящее время нет другого веб-сервера для расчета гидрофобных кластеров и сетей водородных связей. Модульный характер набора инструментов упрощает добавление других приложений в ProteinTools.Мы предполагаем интеграцию приложения для генерации мутантов и оценки их ΔΔG°, а также вычисления полостей в ближайшем будущем. Учитывая текущую потребность в инструментах для анализа растущего числа белковых структур и возможности их расширения, мы твердо верим, что ProteinTools станет незаменимым набором инструментов для сообщества исследователей белков.
Учитывая текущую потребность в инструментах для анализа растущего числа белковых структур и возможности их расширения, мы твердо верим, что ProteinTools станет незаменимым набором инструментов для сообщества исследователей белков.
НАЛИЧИЕ ДАННЫХ
Веб-сервер доступен по адресу https://proteintools.uni-bayreuth.de.
ДОПОЛНИТЕЛЬНЫЕ ДАННЫЕ
Дополнительные данные доступны на сайте NAR Online.
БЛАГОДАРНОСТИ
Мы благодарим сотрудников лаборатории Höcker и Яна Золлера за их подробные отзывы о веб-сайте.
ФИНАНСИРОВАНИЕ
Европейский исследовательский совет [грант ERC Consolidator 647548 «Protein Lego» и грант h3020-FETopen-RIA 764434 «PReART»]; Volkswagenstiftung [94747]. Финансирование платы за открытый доступ: Немецкий исследовательский фонд (DFG) и Университет Байройта в рамках программы финансирования Open Access Publishing, а также ERC [647548].
Заявление о конфликте интересов . Ни один не заявил.
ССЫЛКИ
1.Гринер
Дж.Г.
,Кандатил
С.М.
,Jones
D.T.
Глубокое обучение расширяет покрытие геномов моделированием белков de novo с использованием итеративно прогнозируемых структурных ограничений
.Нац. коммун.
2019
;10
:3977
.2.Старший
А.W.
,Evans
R.
,Jumper
,J.
,KirkPatrick
J.
,Sifre
L.
,Green
T.
,QIN
C.
,Жидек
А.
,Нельсон
AWR
,Bridgland
A.
и др. .Улучшенное предсказание структуры белка с использованием потенциалов глубокого обучения
.Природа
.2020
;577
:706
–710
.3.Lauro
G.
G.
,Ferruz
N.
,Fulle
S.
,Harvery
M.j.
,FINN
P.W.
,De Fabritiis
G.
Переоценка стыковочных поз с использованием молекулярного моделирования и приближенных методов свободной энергии
.J. Chem. Инф. Модель.
2014
;54
:2185
–2189
.4.Lindahl
E.
,Hess
B.
,van der Spoel
D.
пакет для молекулярного анализа и 9 GROMACS 3.0.
Дж. Мол. Модель.
2001
;7
:306
–317
.5.Макгиббон
Р.Т.
,Бошан
К.А.
,Харриган
М.П.
,Кляйн
К.
,Swaels
J.M.
,Hernández
C.X.
,Schwantes
CR
,Wang
LP
,Lane
T.J.
,Панде
В.С.
MDTraj: современная библиотека ppen для анализа траекторий молекулярной динамики
.
Биофиз. Дж.
2015
;109
:1528
–1532
.6.Хаббард
Р.E.
,Камран Хайдер
M.
Водородные связи в белках: роль и сила
.Энциклопедия наук о жизни
.2010
;Чичестер, Великобритания
John Wiley & Sons, Ltd
.7.Йесудас
Дж.П.
,Сайед
Ф.Б.
,Суреш
К.Х.
Анализ взаимодействия структурной воды и CH•••π в протеазе ВИЧ-1 и комплексах PTP1B с использованием инструмента прогнозирования водородных связей, HBPredicT
.Дж. Мол. Модель.
2011
;17
:401
–413
.8.Петтерсен
Э.Ф.
,Годдард
Т.Д.
,Хуанг
К.К.
,Диван
Г.С.
,Гринблатт
Д.М.
,Мэн
ЕС
,Феррин
Т.Е.
UCSF Chimera — система визуализации для поисковых исследований и анализа
.Дж. Вычисл. хим.
2004
;25
:1605
–1612
.10.Humphrey
W.
,Dalke
A.
,Schulten
K.
VMD: визуальная молекулярная динамика
.Дж. Мол. График
1996
;14
:33
–38
.11.Бикади
З.
,Демко
Л.
,Хазай
Э.
Функциональная и структурная характеристика белка на основе анализа его сети водородных связей с помощью графика водородных связей
.Арх. Биохим. Биофиз.
2007
;461
:225
–234
.12.Брэди
Г.П.
,Стоутен
П.Ф.В.
Быстрое прогнозирование и визуализация карманов связывания белков с помощью PASS
.Дж. Вычисл. Помощь. Мол. Дес.
2000
;14
:383
–401
.13.Weisel
M.
,Proschak
E.
,Schneider
G.
PocketPicker: анализ сайтов связывания лигандов 9.
Хим. цент. Дж.
2007
;1
:7
.14.Stourac
j.
,Vavra
O.
O.
,Kokkonen
P.
,FILIPOVIC
J.
,Pinto
G.
,Брезовский
Дж.
,Дамборский
Дж.
,Беднар
Д.
Caver Web 1.0: идентификация туннелей и каналов в белках.
Рез. нуклеиновых кислот.
2019
;47
:W414
–W422
.15.Костантини
С.
,Колонна
Г.
,Факкиано
А.М.
ESBRI: веб-сервер для оценки солевых мостиков в белках
.Биоинформация
.2008
;3
:137
–138
.16.Сен Гупта
P.S.
,Mondal
S.
,Mondal
B.
,Ul Islam
R.N.
,Banerjee
S.
,Bandyopadhyay
А.К.
SBION: программа для анализа солевых мостиков из нескольких структурных файлов
.Биоинформация
.2014
;10
:164
–166
.17.Zimmermann
L.
,Stephens
A.
,Nam
S.Z.
,RAU
D.
,kübler
,j.
,lozajic
M.
,M.
,,
F.
,Södd
J.
,Lupas
A.N.
,Alva
V.
Полностью переработанный инструментарий биоинформатики MPI с новым сервером HHpred в основе
.Дж. Мол. биол.
2018
;430
:2237
–2243
.18.Martínez-Rosell
G.
,Giorgino
,Giorgino
T.
,DE FABRITIIS
G.
G.
PlayMolecule Playmolecule ProteinPrepare: веб-приложение для препарата белка для моделирования молекулярной динамики
.J. Chem. Инф. Модель.
2017
;57
:1511
–1516
.19.Копыта
Р.В.В.
,Vriend
G.
,Sander
C.
,Abola
E.E.
Ошибки в белковых структурах [3]
.Природа
.1996
;381
:272
.20.Дэвис
И.В.
,Блок
Дж.Н.
,Капрал
Г.Дж.
,Ван
Х.
,Мюррей
Л.В.
,Арендал
В.Б.
,Сноеинк
Дж.
,Ричардсон
Дж.С.
и др. .MolProbity: всеатомные контакты и проверка структуры белков и нуклеиновых кислот
.Рез. нуклеиновых кислот.
2007
;35
:375
–383
.21.Джерат
Г.
,Хазам
П.К.
,Рамакришнан
В.
Набор инструментов bPE: набор инструментов для вычислительной белковой инженерии
.Сист. Синтез. биол.
2014
;8
:337
–341
.22.Соболев
V.
,Сорокин
A.
,Prilusky
J.
,ABola
E.E.
,EDELMAN
M.
M.
Автоматизированный анализ межатомных контактов в белках
.Биоинформатика
.1999
;15
:327
–332
.23.Катурия
С.В.
,Чан
Ю.Х.
,Нобрега
Р.П.
,Matthews
C.R.
Кластеры боковых цепей изолейцина, лейцина и валина определяют ядра стабильности в высокоэнергетических состояниях глобулярных белков: последовательности, определяющие структуру и стабильность
.Науки о белках.
2016
;25
:662
–675
.24.Lechner
H.
,Ferruz
N.
,Höcker
B.
Стратегии создания ненатуральных ферментов и связующих18.
Курс. мнение хим. биол.
2018
;47
:67
–76
.25.Basak
S.
,Paul Nobrega
R.
,TAVELLA
D.
,DEVEA
LM
,Koga
N.
,Tatsumi-Koga
R.
,Baker
D.
,Massi
F.
,Robert Matthews
C.
Сети электростатических и гидрофобных взаимодействий модулируют сложную складчатую поверхность свободной энергии разработанного белка βα
.Проц. Натл. акад. науч. США
2019
;116
:6806
–6811
.26.KOGA
N.
N.
,Tatsumi-Koga
R
,LIU
G.
,Xiao
R
,Acton
T.B.
,Монтелионе
Г.Т.
,Baker
D.
Принципы проектирования идеальных белковых структур
.Природа
.2012
;491
:222
–227
.27.Каллебаут
И.
,Labesse
G.
,Durand
,P.
,Poupon
A.
,,
,
L.
,Chomilier
J.
,Henrissat
B.
,Mornon
JP
Расшифровка информации о последовательности белка с помощью гидрофобного кластерного анализа (HCA): текущее состояние и перспективы
.Сотовый. Мол. Жизнь наук.
1997
;53
:621
–645
.28.Bitard-Feildel
T.
,Callebaut
I.
HCAtk и pyHCA: набор инструментов и Python API для гидрофобного кластерного анализа белковых последовательностей
.2018
; https://doi.org/10.1101/249995.29.Соболев
В.
,Уэйд
Р.К.
,Vriend
G.
,Edelman
M.
Молекулярный докинг с использованием поверхностной комплементарности
.Структура белков. Функц. Биоинформа.
1996
;25
:120
–129
.30.Соболев
В.
,Эдельман
М.
Моделирование сайта связывания хинона-В реакционного центра фотосистемы-II с использованием представлений о комплементарности и поверхности контакта между атомами
.Структура белков. Функц. Биоинформа.
1995
;21
:214
–225
.31.Shannon
R.D.
Пересмотренные эффективные ионные радиусы и систематические исследования межатомных расстояний в галогенидах и халькогенидах
.Акта Кристаллогр. Разд. А
.1976
;32
:751
–767
.32.Волек
К.
,Гомес-Сицилия
À.
,Циеплак
М.
Определение карт контактов в белках: сочетание структурного и химического подходов
.J. Chem. физ.
2015
;143
:243105
.33.Гонсалес
А.
Измерение площадей на сфере с использованием Фибоначчи и решеток широты-долготы
.Матем. Geosci.
2010
;42
:49
–64
.34.Олссон
М.Х.М.
,Søndergaard
C.R.
,Rostkowski
M.
,Jensen
J.H.
PROPKA3: последовательная обработка внутренних и поверхностных остатков в эмпирических предсказаниях p K a
.J. Chem. Теория вычисл.
2011
;7
:525
–537
.35.Долинский
Т.Дж.
,Nielsen
J.E.
,McCammon
J.A.
,Baker
Н.Д.
PDB2PQR: автоматизированный конвейер для настройки электростатических расчетов Пуассона-Больцмана
.Рез. нуклеиновых кислот.
2004
;32
:665
–667
.36.Бейкер
E.N.
,Хаббард
Р.Э.
Водородные связи в глобулярных белках
.Прог. Биофиз. Мол. биол.
1984
;44
:97
–179
.37.Дас
Р.К.
,Паппу
Р.В.
На конформации внутренне неупорядоченных белков влияет линейное распределение последовательностей противоположно заряженных остатков
.Проц. Натл. акад. науч. США
.2013
;110
:13392
–13397
.38.Дырка
А.С.
,Дас
Р.К.
,Ахад
Дж.Н.
,Ричардсон
М.О.Г.
,Паппу
Р.В.
CIDER: ресурсы для анализа взаимосвязей последовательностей и ансамблей внутренне неупорядоченных белков
.Биофиз. Дж.
2017
;112
:16
–21
.39.Мир
С.
,Альхруб
Ю.
,Аньянго
С.
,Армстронг
Д.Р.
,Беррисфорд
Дж. М.
,Кларк
А.Р.
,Конрой
М.Дж.
,Дана
Дж.М. .
PDBe: к многоразовой инфраструктуре доставки данных в банке белковых данных в Европе
.Рез. нуклеиновых кислот.
2018
;46
:D486
–D492
.40.Фокс
Н.К.
,Бреннер
ЮВ
,Чандония
Ж.-М.
SCOPe: структурная классификация белков – расширенная, объединяющая данные SCOP и ASTRAL и классификация новых структур
.Рез. нуклеиновых кислот.
2014
;42
:D304
–D309
.41.Vassura
M.
M.
,Margara
L.
,Di Lena
P.
,Medri
F.
,Fariselli
P.
,Casadio
R.
Реконструкция трехмерных структур по картам контактов белков
.IEEE/ACM Transactions по вычислительной биологии и биоинформатике
.2008
;5
:357
–367
.42.Холм
Л.
DALI и сохранение формы белка
.Науки о белках.
2020
;29
:128
–140
.43.Ferruz
N.
N.
,LOBOS
F.
,LEMM
D.
,TOLEDO-PATINO
S.
,Farías-Rico
J.A.
,Schmidt
S.
,Höcker
B.
Идентификация и анализ природных строительных блоков для направленной эволюции конструкции белков на основе фрагментов
.Дж. Мол. биол.
2020
;432
:3898
–3914
.© Автор(ы), 2021 г. Опубликовано Oxford University Press от имени Nucleic Acids Research.
Это статья в открытом доступе, распространяемая в соответствии с лицензией Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), которая разрешает неограниченное повторное использование, распространение и воспроизведение на любом носителе при условии, что оригинал работа цитируется правильно.| Малоугловое рассеяние рентгеновских лучей (SAXS) | ||||
| Дифракцию на некристаллических образцах, представляющих собой порошки или растворы, в которых все молекулы ориентированы случайным образом, обычно называют рассеянием. Дифракционная картина усредняется по всем направлениям, сферически, потому что рентгеновский луч сталкивается со всеми возможными ориентациями молекул в образце. Дифракционная картина по-прежнему содержит информацию о том, как плотность электронов меняется с расстоянием от центра молекул, составляющих этот образец. | Анализ интенсивностей под разными углами рентгеновского излучения к образцу (маленький) дает функцию распределения расстояний, которая дает частоты всех возможных внутримолекулярных расстояний в белке. Из этого вы можете смоделировать общую форму белка и создать простую белковую оболочку. Поскольку образцы находятся в растворе, вы можете легко обнаружить динамику, связывание и конформационные изменения. Данные также позволяют рассчитать радиус вращения (расстояние, на которое распространяется масса). | Данные могут быть записаны относительно быстро за 1 день, и для вычета вклада буфера в рассеяние требуются только хорошо подобранные буферные растворы.Некоторые учреждения теперь могут анализировать несколько образцов в 96-луночном планшете, но чаще всего одновременно можно измерять только один образец. | ||
| Изотермическое титрование Калориметрия | ||||
| Измерение изменения температуры при добавлении молекул к белковым растворам. | Он показывает, насколько хорошо молекула связывается, а также энтальпию, энтропию и свободную энергию взаимодействия. | Одно титрование для одного взаимодействия занимает приблизительно 2 часа. | ||
| Собственная масс-спектрометрия | ||||
| Электрораспылительная ионизация работает при пропускании тока через летучий растворитель.Это приводит к тому, что белковые комплексы ионизируются и переходят в газовую фазу. Молекулярную массу можно рассчитать по тому, сколько времени требуется ионам, чтобы пройти заданное расстояние. Это называется временем пролета (TOF). | Молекулярную массу белков и комплексов можно определить в газовой фазе. Его можно использовать, чтобы сказать вам через изменения массы, связан ли белок с другой молекулой, например, ионом металла или лекарством. | Каждый спектр можно получить за несколько секунд, поэтому за день можно измерить множество образцов, но анализ данных может занять гораздо больше часов. | ||
| Общая флуоресценция | ||||
| Флуоресценция включает использование луча света, который возбуждает электроны в молекулах определенных соединений и заставляет их излучать свет с большей длиной волны. | Различные флуорофоры поглощают и излучают свет с разной длиной волны в зависимости от их локального окружения. Например, молекула 8-анилино-1-нафталинсульфоновой кислоты (ANS) является широко используемым флуоресцентным зондом для характеристики белков и сайтов связывания, поскольку она флуоресцирует только при связывании с гидрофобными пятнами на белке. | Процесс очень быстрый, занимает миллисекунды. С помощью многолуночных планшет-ридеров можно записать многие сотни измерений в течение нескольких минут. | ||
| Дифференциальная сканирующая флуориметрия | ||||
| Когда белки свернуты, они прячут свое гидрофобное ядро и не могут связывать флуоресцентный краситель. Используя тепло во время линейного изменения температуры, белок разворачивается и связывает краситель, и флуоресценция красителя увеличивается. | Указывает температуру, при которой разворачивается половина белка, также известную как T m .Если вы добавите молекулу лекарства к белку, T m увеличится, и это может сказать вам, насколько хорошо оно связывается. | Вы можете измерить 96 образцов всего за 1 час. Он очень популярен для проверки многих партнеров по связыванию и буферных условий. | ||
| Внутренняя флуоресценция триптофана | ||||
| Внутри белков аминокислотная боковая цепь триптофана флуоресцирует. Длина волны излучаемого света колеблется от примерно 300 нм в неполярных средах, таких как внутренняя часть белка, до 350 нм в водной полярной среде на поверхности. | Поскольку пиковая длина волны излучаемого света зависит от окружающей среды вокруг боковой цепи аминокислоты, флуоресценция может использоваться в качестве очень чувствительного измерения конформационного состояния отдельных остатков триптофана. Если излучаемый свет ближе к 300 нм, то триптофан находится в неполярной среде, а если он ближе к 350 нм, он находится в водной полярной среде. | Как и при общей флуоресценции, процесс очень быстрый. Как правило, спектры излучения можно получить менее чем за минуту, что означает возможность быстрого анализа многих образцов. | ||
| Химическая денатурация с последующей внутренней флуоресценцией триптофана Эти химические вещества титруют раствором свернутого белка и измеряют флуоресценцию в каждой точке. | Данные наносятся на график и составляют кривую денатурации, которая показывает концентрацию денатуранта, при которой разворачивается половина белка.Наклон перехода также говорит вам, насколько белок чувствителен к денатуранту. Вместе эти значения позволяют рассчитать изменение свободной энергии для разворачивания, что является абсолютной мерой стабильности белка. Если в отдельный эксперимент также включены лекарственные препараты или лиганды, то можно рассчитать константу связывания. Белки также можно внезапно заставить свернуть или развернуть, при этом изменение флуоресценции можно измерить в режиме реального времени, чтобы понять кинетику сворачивания белка. | Эти анализы обычно проводят в 96-луночном планшете с использованием небольших объемов и низких концентраций белков.Титрование всего планшета может занять около 6 часов и требует больше анализа данных, чем дифференциальная сканирующая флуориметрия, но дает более точные и количественные данные. Кинетика разворачивания белка также может быть выполнена в планшете, но прямая кинетика складывания обычно требует спектрометра с остановленным потоком и имеет меньшую производительность. | ||
| Резонансный перенос энергии флуоресценции | ||||
| Резонансный перенос энергии флуоресценции (FRET) — это зависящий от расстояния физический процесс, посредством которого энергия передается между двумя флуорофорами.Свет поглощается флуорофором на одной длине волны (возбуждение), после чего следует излучение с большей длиной волны, которое поглощается соседним флуорофором, который затем испускает обнаруженный свет с еще большей длиной волны. В идеале эти флуорофоры должны иметь узкие, но частично перекрывающиеся линии излучения. Пара FRET может представлять собой небольшие молекулы, такие как родамин и флуоресцеин, которые перекрестно связаны непосредственно с белком. В качестве альтернативы такие молекулы, как зеленый или синий флуоресцентный белок (GFP/BFP), могут быть связаны непосредственно с концами двух исследуемых белков. | Можно использовать в качестве молекулярной линейки, чтобы определить, насколько близко друг к другу находятся две молекулы. Один белок помечен донорным флуорофором, а второй – акцепторным флуорофором. Если они находятся в пределах нескольких нанометров, то происходит передача энергии. Он используется для измерения динамики и белковых взаимодействий. | Получение данных для FRET происходит быстро, как только белки помечены. Однако присоединение флуоресцентных зондов к белку может занять много часов или дней. | ||
| Вычислительная биология белков | ||||
| Вычислительная биология может использоваться для прогнозирования структуры и динамики белков.Было разработано много мощных алгоритмов, которые учитывают химические свойства аминокислотной последовательности для характеристики белков. Моделирование гомологии использует последовательность белка с неизвестной структурой с известной структурой, которая обычно находится в родственном семействе (см. SCOP и CATH) для моделирования неизвестной структуры. Этот метод является активной областью исследований. Другой важной областью является моделирование молекулярной динамики, которое применяет к белкам правила химии и физики, определяющие поведение молекул в водной среде.Общесистемный анализ белков использует протеом организма, который представляет собой все его белковые последовательности, определенные в результате секвенирования генома. | Эти методы создают несколько важных баз данных белков с предсказанными структурами, взаимодействиями и эволюционными отношениями, которые позволяют генерировать гипотезы, которые можно проверить в лаборатории. Молекулярная динамика создает фильмы о движении белков, которые предоставляют новую информацию о поведении белков, которую нельзя было увидеть с помощью традиционных экспериментальных методов.Новая область системной биологии пытается объединить всю доступную информацию о белках в организме, чтобы смоделировать, как все они работают вместе в клетке или организме в целом, чтобы выполнять жизненные функции. | Многие базы данных делают предсказания о белках автоматически, и информация легко доступна для всех. Например, база данных UniProt содержит всеобъемлющий, высококачественный и свободно доступный ресурс информации о последовательностях и функциях белков. Моделирование молекулярной динамики может занять много дней, чтобы выполнить и проанализировать, и в настоящее время мы ограничены просмотром только микросекунд движения.В настоящее время подходы системной биологии также требуют очень много времени. Однако вычислительные методы могут сэкономить много времени и усилий, поскольку компьютеры являются дешевыми, но мощными инструментами, которые можно использовать в сочетании с экспериментальными методами. | ||
Четыре уровня белковой структуры – клеточная физиология
Структура и функция белков
Белки являются строительными блоками клеточных структур и двигателями клеточной активности. Они имеют модульную природу, и их взаимодействие с другими молекулами в клетке зависит от наличия специфических функциональных доменов.Точная форма домена, обусловленная наличием нековалентных связей между остатками в полипептидной цепи, определяет функцию. Наиболее известным примером взаимосвязи форма-функция является теория ферментативной функции «ключ-замок». Изменение ферментативного кармана вследствие мутации или модификации аминокислотного остатка изменяет аффинность и/или специфичность фермента. Короче говоря, чем лучше подходят две молекулы, тем лучше они функционируют, тем больше связей может быть образовано, тем быстрее может передаваться сигнал или тем сильнее соединяются две молекулы (вспомните молекулы адгезии).
Трехмерная конформация белка зависит от взаимодействия между аминокислотами в полипептидной цепи. Поскольку последовательность аминокислот зависит от генетического кода, форма белка закодирована в ДНК. Белки имеют четыре уровня организации. Первичная структура относится к линейной последовательности аминокислот, соединенных пептидными связями. Вторичная структура состоит из локальной упаковки полипептидной цепи в α-спирали и β-слои за счет водородных связей между пептидной связью и центральным углеродным остовом. Третичная (3D) структура представляет собой форму, возникающую в результате складывания вторичных структур, определяемых взаимодействиями между боковыми цепями аминокислот. Четвертичная структура описывает расположение полипептидных цепей в многосубъединичной структуре.
В этом видео показаны 4 уровня структуры белка.
Взято из банка данных RCBProtein по лицензии CC-BY
Все, что необходимо для придания белку уникальной формы и, следовательно, уникальной функции, «записано» во фрагменте ДНК, известном как ген.Каждый раз, когда транскрибируется ген, либо в течение жизни клетки, либо в любой клетке, имеющей одинаковую ДНК, природную или рекомбинантную, белки появляются одинаково и принимают на себя заранее запрограммированную функцию.
Первичная структура белков
Белки являются наиболее важным и универсальным классом макромолекул в клетке. Роли, которые играют эти молекулы, охватывают все: от транспортировки питательных веществ, катализа биохимических реакций до структурных компонентов клеток или молекулярных двигателей.Белки представляют собой линейные полимеры аминокислот, соединенных пептидными связями. Они синтезируются из матричной нити ДНК и содержат уникальные и специфические аминокислотные последовательности в линейной форме, известной как первичная структура .
Всего двадцать аминокислот необходимы и достаточны для образования тысяч белков в клетке. Это не значит, что аминокислот всего двадцать. Это распространенное заблуждение. В мире существует бесчисленное множество аминокислот, но они участвуют в других метаболических реакциях, но не в синтезе белка.То, как индивидуальный белок обретает свою идентичность, зависит от упорядоченной комбинации аминокислот, которая определяет все его характеристики.
Аминокислоты, соединенные пептидной связью, называются полипептидной цепью. Полипептидная цепь состоит из последовательностей аминокислот, продиктованных геном. Последовательность аминокислотных цепей обеспечивает разнообразие, необходимое для удовлетворения потребностей жизни. Сохранение специфических белковых последовательностей настолько важно, что в клетке есть регулирующие механизмы, гарантирующие производство только идеальных белков.Каждая отдельная последовательность имеет уникальный порядок, который передает очень уникальную функцию. Если бы вы изменили одно-единственное расположение цепи, то у этой цепи была бы совершенно другая функция. Функция белка может быть нарушена или полностью потеряна, если последовательность нарушена. Но не все мутации или модификации белков приводят к катастрофическим последствиям. Некоторые из них заставляют клетку и организм лучше приспосабливаться к давлению окружающей среды, процесс, известный вам как эволюция.
Свойства аминокислот и различия их боковых цепейАминокислоты имеют одинаковую базовую структуру, что важно для образования правильной химической связи между соседними молекулами.Каждая аминокислота имеет центральный углерод, обозначаемый как α-углерод. К α-углероду всегда присоединены следующие четыре группы:
- –Nh3 основная аминогруппа
- –COOH кислотная группа (известная как карбоксильная группа)
- –H атом водорода
- –R боковая цепь
-R символизирует вариабельную боковую цепь, которая является единственной химической группой , отличающейся среди всех двадцати аминокислот. По сути, боковая цепь делает аминокислоту уникальной, и ее можно рассматривать как ее отпечаток пальца.
Важнейшим свойством аминокислот, влияющим на сворачивание, а впоследствии и на функцию всей белковой молекулы, является их известное и предсказуемое взаимодействие с водой. Следовательно, аминокислоты можно разделить на гидрофильные и гидрофобные группы. Гидрофобные, или неполярные, аминокислоты имеют насыщенные углеводороды в качестве боковых цепей. Этими аминокислотами являются аланин, валин, метионин, лейцин и изолейцин и две аминокислоты с ароматическими кольцами триптофан и фенилаланин .Гидрофобные, неполярные аминокислоты играют важную роль в сворачивании белков, потому что они склонны сближаться и слипаться от воды. Эти аминокислоты обычно образуют трансмембранные домены и обнаруживаются глубоко скрытыми в гидрофобной внутренней части большинства глобулярных белков.
Гидрофильные аминокислоты легко взаимодействуют с водой. В эту группу входят аминокислоты, которые ионизируются и приобретают электрический заряд (как отрицательный, так и положительный) при диссоциации, а также полярные, но незаряженные аминокислоты.Аминокислоты, которые имеют боковые цепи с карбоксильной группой в дополнение к карбоксильной группе при α-углероде, используемом при образовании пептидной связи, несут отрицательный заряд. Этими остатками являются глутаминовая кислота и аспарагиновая кислота — обратите внимание, что их названия фактически содержат термин «кислота» из-за наличия ДВУХ карбоксильных групп.
Боковые цепи лизина, аргинина и гистидина имеют сильные основные группы и положительно заряжены. Гидрофильные аминокислоты, которые являются полярными, но незаряженными, представляют собой аспарагин, глутамин, серин, треонин и тирозин .Гидрофильные и заряженные боковые цепи аминокислот экспонированы на поверхности белка и особенно широко распространены в ферментативных карманах или транспортных молекулах. Открытые электрические заряды передают природу и активность белка другим молекулам и действуют как магниты, притягивающие аналогичные силы для взаимодействия.
Некоторые аминокислоты вносят вклад в структуру белка из-за уникальных особенностей, характерных для их боковых цепей. Структура пролина отличается от других аминокислот тем, что его боковая цепь связана с азотом, а также с центральным углеродом.Эта аминокислота химически нереактивна (гидрофобна), но из-за своего пятичленного кольца она нарушает геометрию сворачивающегося белка, вызывая резкие сдвиги в конформации, физически вводя перегибы и изгибы в полипептидную цепь. Глицин вообще не имеет боковой цепи, только второй атом водорода, присоединенный к α-углероду. Не проявляя сильного полярного характера или электроотрицательности, он обычно наблюдается в местах, где части полипептидной цепи изгибаются и сближаются друг с другом.
Цистеин представляет собой аминокислоту, широко известную тем, что она сильно влияет на структуру белка . У него есть сульфгидрильная группа, ответственная за образование дисульфидных связей, которые стабилизируют третичную структуру белков и вносят большой вклад в молекулярные функции, о которых вы узнаете позже в этом тексте.
Вторичная структура и все петли
Откуда мы знаем, как на самом деле выглядят белки, когда они свернуты? Есть два метода, позволяющих заглянуть в структуру белка; рентгеновская дифракция и ядерный магнитный резонанс (ЯМР).Метод рентгеновской дифракции создает трехмерную контурную карту электронов в белковом кристалле на основе того, как рентгеновские лучи отражаются при прохождении через образец. ЯМР измеряет расстояние между белками в насыщенном растворе, и информация о пространственных ограничениях используется для определения складчатых структур каждого белка. Эти два теста вместе помогают нам понять, какова свернутая форма белка.
Форма белка определяется исключительно последовательностью аминокислот в полипептидной цепи.Верно; это точно так же, как ДНК, уникальный код создает уникальный дизайн. Сворачивание белков является результатом физических свойств боковых цепей аминокислот и их взаимодействия с окружающей средой. Белки сворачиваются в наиболее энергоэффективную форму, называемую нативным состоянием , в несколько этапов или уровней в структуре белка.
Фолдинг и архитектура белковПри воздействии условий в цитозоле или просвете ER полипептидные цепи приобретают локализованную организацию, называемую вторичной структурой , которая оптимизирует взаимодействие между боковыми цепями аминокислот друг с другом и водой.Полипептидный остов сворачивается в спирали и ленты соответственно α-спиралей и β-листов. Как α-спираль, так и β-лист представляют собой сегменты полипептида, имеющие правильную геометрию, сплетенные между собой пологими и не очень пологими поворотами и разделенные менее организованными петлями.
Альфа-спиральпредставляет собой структуру, которая упаковывает α-углероды с вращением, обеспечивающим благоприятные углы для образования прочных водородных связей и плотной упаковки боковых цепей. Бета-листы представляют собой плоские структуры, состоящие из нескольких β-тяжей, связанных с соседними β-тяжами посредством водородных связей.В β-листах полипептидная цепь может идти в одном (параллельном) или противоположном направлении (антипараллельном). Водородные связи более стабильны, когда β-лист имеет антипараллельные, а не параллельные нити. Параллельные слои, как правило, скрыты внутри белковой структуры. Вторичные структуры соединены неструктурированными участками, образующими несколько петель.
Третичная структура белка
Существует множество способов объединения вторичных структур в большую трехмерную решетку. Третичная структура белка представляет собой трехмерную комбинацию α-спиралей и β-листов, которые складываются рядом друг с другом в результате нековалентных взаимодействий между боковыми группами аминокислот и окружением, окружающим отдельный полипептид. На этой стадии белки начинают укреплять свою структуру за счет дополнительных связей, таких как дисульфидные связи между двумя цистеинами . Наиболее важной особенностью третичных структур является наличие консервативных областей со схожими функциями, известных как функциональных доменов .Третичные структуры менее стабильны, и действительно, большинство из них меняют форму в течение жизни белка, часто многократно. Конформационные изменения внутри этих функциональных доменов являются основой функции белка. Они могут быть постоянными в процессе фолдинга и созревания белка или обратимыми и служить способом регуляции активности белка в реакции по шкале реакций. Белковые домены представляют собой области с аналогичной активностью. Они не обязательно имеют консервативную последовательность. Например, домен киназы, ответственный за присоединение фосфатной группы, имеет различную форму и последовательность в зависимости от субстрата, к которому присоединена фосфатная группа.Вторичные структуры, образующие домены, не обязательно должны лежать последовательно в полипептидной цепи. Они могут даже быть частями нескольких разных полипептидов в случае мультимерных белков.
Мотивы представляют собой подгруппу функциональных доменов, которые имеют эволюционно консервативные последовательности , придающие им, разумеется, консервативную форму. Один пример, спирально-спиральные мотивы представляют собой очень регулярные суперструктуры двух α-спиралей, соединенных в пары, чтобы сформировать волокнистую конфигурацию, которая является основой стабильных димеров.Обычно имеются две идентичные α-спирали, закрученные друг вокруг друга в левой конформации и стабилизированные гидрофобными взаимодействиями. Межмолекулярные ионные связи между боковыми цепями в α-спирали, разделенные 3,6 остатками, дают гидрофобным остаткам пространство для взаимодействия с аналогичным мотивом на противоположном белке.
Четвертичная структура
Четвертичная структура является результатом сборки двух или более полипептидов в один функциональный мультимерный белок .Субъединицы собираются за счет взаимодействий между доменами или областями белка и удерживаются вместе гидрофобными взаимодействиями (два влажных зеркала) и дисульфидными связями. Если субъединицы одинаковые, структура описывается префиксом гомо , а если они различаются префиксом гетеро (как в мышечном гликогенфосфорилазе гомодимер или как в гетеротримерных G белках)
Внутриклеточные процессы, такие как передача сигналов, зависят от взаимодействия между молекулами.Чем лучше молекулярное соответствие между двумя молекулами, тем больше связей они могут образовать или тем сильнее взаимодействие (сродство между ними). Последовательность аминокислот определяется геном, и, в свою очередь, свойства боковых цепей аминокислот определяют форму и, в свою очередь, взаимодействие.
.